Article Text

Abstract
Background and purpose C-C motif chemokine ligand 17 (CCL17) presents an important role in immune regulation, which is critical in the pathophysiology of brain injury after subarachnoid haemorrhage (SAH). There is rare evidence to illustrate the function of CCL17 towards SAH. In this study, we try to reveal the therapeutic effects of CCL17 and its underlying mechanism in rat SAH model.
Methods SAH rat models were assigned to receive recombinant CCL17 (rCCL17) or phosphate buffer saline (PBS). AZD2098 and JR-AB2-011 were applied to investigate the C-C motif chemokine receptor 4 (CCR4)/mammalian target of rapamycin complex 2 (mTORC2) axis in CCL17-mediated neuroprotection. To elucidate the underlying mechanism, the in vitro kinase assay was performed in primary microglia. Microglial-specific Rictor knockdown was administered via intracerebroventricular injection of adenovirus-associated virus. Brain water content, short-term neurobehavioural evaluation, western blot analysis, quantitative RT-PCR and histological staining were performed.
Results The expression of CCL17 was increased and secreted from neurons after oxyhaemoglobin stimulation. Exogenous rCCL17 significantly alleviated neuronal apoptosis, and alleviated short-term neurofunction after SAH in rats. In addition, rCCL17 increased M2-like polarisation of microglia in rats post-SAH and in primary microglia culture. The neuroprotection of rCCL17 was abolished via inhibition of either CCR4 or mTORC2.
Conclusion CCL17 activated the CCR4/mTORC2 axis in microglia, which can alleviate SAH-induced neurological deficits by promoting M2-like polarisation of microglia.
- subarachnoid
- brain
- cerebrovascular disorders
- hemorrhage
Data availability statement
Data are available on reasonable request. All raw data used in this manuscript are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Endogenous C-C motif chemokine ligand 17 (CCL17) was proved to be dominantly expressed in hippocampal neuron.
Additionally, exogenous CCL17 can enhance haematoma resolution in mice intracerebral haemorrhage models; however, its neuroprotective role and the underlying mechanisms of CCL17 after subarachnoid haemorrhage (SAH) onset was still not yet unravelled.
WHAT THIS STUDY ADDS
Exogenous CCL17 treatment improved early brain injury after SAH and promotes M2-like polarisation of microglia through the C-C motif chemokine receptor 4 (CCR4)/mammalian target of rapamycin complex 2 (mTORC2) signal pathway in rats.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
We demonstrated that administration of recombinant CCL17 can alleviate nervous system lesions, at least in part, through the CCR4/mTORC2 signalling pathway, and improved outcomes after SAH, which emphasised the potential therapeutic values of CCL17 in future clinical management.
Introduction
Subarachnoid haemorrhage (SAH), as a severe and devastating cerebrovascular disease, has a high rate of mortality and morbidity.1–3 Of note, nearly half of the survivors suffer from severe neurological dysfunction, which introduces a substantial burden to societal and familial burdens.2 4 Currently, researchers have proposed that the ‘early brain injury’ (EBI) may be an essential factor for neurological deficits after SAH.5 However, lacking effective pharmaceutical strategy for EBI made it a predominant challenge in clinical therapy.6 7
Microglia, as innate immune cells, are responsible for the early inflammation after SAH.7–9 Accumulating evidence has revealed that microglial polarisation to selectively activate microglial M2 phenotypes to assist in tissue repair under a variety of conditions, such as ischaemic stroke,10 traumatic brain injury11 and intracerebral haemorrhage (ICH).12 M2 microglia can promote haematoma clearance and neuronal survival, protect neurobehavioural function and attenuate apoptosis.13–16 Therefore, it is of utmost importance to elucidate the regulation of M2 microglia infiltration in the brain after SAH.
CCL17 is known to participate in trafficking immune cells.17 In the central nervous system, CCL17 is reportedly expressed in hippocampal neurons in murine. Meanwhile, its specific receptor CCR4 was expressed on neurons, astrocytes and microglia.18 19 CCR4-deficient mice displayed distinct behavioural deficits, indicating its possible involvement in regulating neuronal or glial functions.20 21 Besides, we previously reported that CCR4 mediated the activation of the mTOR signal pathway in pituitary adenoma.22 However, the biological function of CCL17/CCR4 and the underlying mechanism of microglial regulation has not been systemically investigated in SAH.
In this study, we hypothesised that CCL17 might attenuate EBI by regulating microglia polarisation through CCR4, and may be a novel therapeutic strategy for SAH. Therefore, we investigated the therapeutic effects of recombinant CCL17 (rCCL17) administration and its underlying mechanism after SAH in a rat model.
Methods
Experimental design
Online supplemental figure 1 provides an overview of the following four experiments and experimental groups. Assessment of images and behavioural scores were performed in a blinded manner.
Supplemental material
Experiment 1: time course of endogenous CCL17 and CCR4 and their cellular localisation after SAH
In this section, 24 rats were randomly assigned to 6 groups: sham, 3 hours, 6 hours, 12 hours, 24 hours, 72 hours post-SAH. The expression of endogenous CCL17 and CCR4 after SAH was detected (n=3). Additional rats were used for immunofluorescence (IF) staining in sham and 24 hours groups (n=3). The extent of CCR4 expression was explored in primary neurons, astrocytes and microglia of rats in vitro after treatment of oxyhaemoglobin (OxyHb).
Experiment 2: the effects of rCCL17 treatment on EBI after SAH
To determine the appropriate treatment dosage for rCCL17, a total of 95 rats were assigned to 5 groups: sham, SAH+vehicle, SAH+rCCL17 (10μg/kg), SAH+rCCL17 (30 μg/kg), or SAH+rCCL17 (60μg/kg). Modified Garcia scores, forelimb placement tests and brain water content (BWC) were assessed at 24 hours and 72 hours after SAH (samples were shared, n=6). IF staining (n=3) and quantitative PCR (qPCR) (n=4) were performed. The efficacy of rCCL17’s function on EBI and microglial morphology was detected at 24 hours post-SAH. The detailed PCR primers are listed in online supplemental table 1.
Experiment 3: the role of the CCR4/mTORC2 signalling pathway post-SAH
To investigate the function of rCCL17 and CCR4 after SAH, 102 rats were divided into 6 groups: sham, SAH+vehicle, SAH+rCCL17 (60 μg/kg), SAH+rCCL17+DMSO, SAH+rCCL17+AZD2098 (1 mg/kg) and SAH+rCCL17+JR-AB2-011 (1 mg/kg). Activation of mTORC2 and ERK signalling was examined (n=3) and the M2-like microglia biomarkers were examined by qPCR (n=4). Neurobehavioural functions and BWC were evaluated at 24 hours and 72 hours after SAH (samples were shared, n=5/group/time point). IF staining (n=3) and qPCR (n=4) were performed. To explore the efficacy of rCCL17 on EBI and microglial morphology, TdT-mediated dUTP nick end labeling (TUNEL), Fluoro-Jade C (FJC) staining and IF were performed at 24 hours post-SAH (n=3).
Experiment 4: the role of CCL17/CCR4/mTORC2 axis in microglia post-SAH
In this experiment, we investigated the effects of rCCL17-regulated microglia after SAH. A total of 102 rats were divided into 6 groups: sham+AAV-CD68-Control, SAH+vehicle+AAV-CD68-Control, SAH+rCCL17+AAV-CD68-Control, sham+AAV-CD68-shRictor, SAH+vehicle+AAV-CD68-shRictor and SAH+rCCL17+AAV-CD68-shRictor. Adeno-associated virus (AAV) was administered via intracerebroventricular injection 21 days before SAH induction. The activation of mTORC2 and ERK signalling was examined (n=3) and the M2-like microglia biomarkers were examined by qPCR (n=4). Neurobehavioural functions and BWC were evaluated at 24 hours and 72 hours after SAH (samples were shared, n=5/group/time point). IF staining (n=3) and qPCR (n=4) were performed. To explore the efficacy of rCCL17 on EBI and microglial morphology, TUNEL, FJC staining and IF were performed at 24 hours post-SAH (n=3). The regulatory function of CCL17 on M2-like polarisation of microglia was demonstrated in rat primary microglia in vitro.
Statistical analysis
SPSS V.22.0 (IBM, USA) or Prism V.9 software program (GraphPad Software, USA) was used for statistical analysis. The normality of data was examined by Shapiro-Wilk test (Gaussian distribution). Based on the distribution of normality, multiple groups were compared via one-way analysis of variance with Tukey’s post hoc test. The difference of data in Gaussian distribution was evaluated using unpaired (two-sided) Student’s t-test, while the difference of data in non-Gaussian distribution was evaluated with Kruskal-Wallis test. Statistically significant differences were considered when p values were <0.05. For in vitro study, each experiment was repeated three times independently with similar results.
Results
Animal use
In this study, a total of 323 rats were used and the mortality of SAH rats was 14.0% (37/264), but no rats died in the sham group (59 rats) (online supplemental figure 1).
CCL17 expression was enhanced in neuron after acute SAH
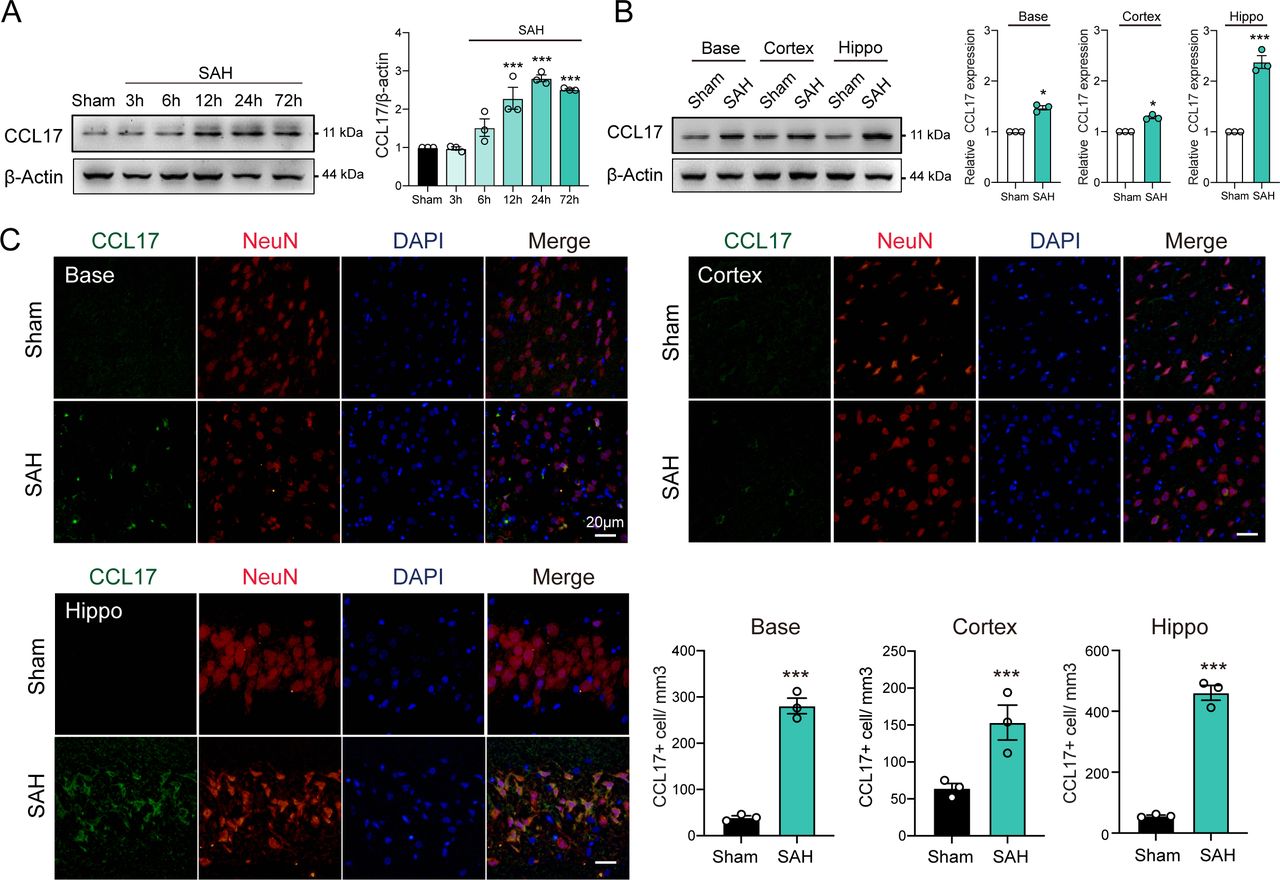
To investigate the dynamic changes of CCL17 expression, we performed western blot analysis of the left hemisphere of rat models from sham, 3 hours, 6 hours, 12 hours, 24 hours and 72 hours post-SAH. The level of endogenous CCL17 was markedly increased at 12 hours, peaked at 24 hours and then decreased at day 7 after SAH (figure 1A). In addition, IF further revealed increased neuronal CCL17 expression in the basal ganglia, temporal cortex and hippocampus at 24 hours after SAH (figure 1B). CCL17 levels of the hippocampus after SAH were significantly higher than the basal ganglia and temporal cortex regions (figure 1B–C). Furthermore, CCL17 expression was greatly higher in the dentate gyrus, as well as in the CA1 and CA3 regions following SAH when compared with sham group (online supplemental figure 2).
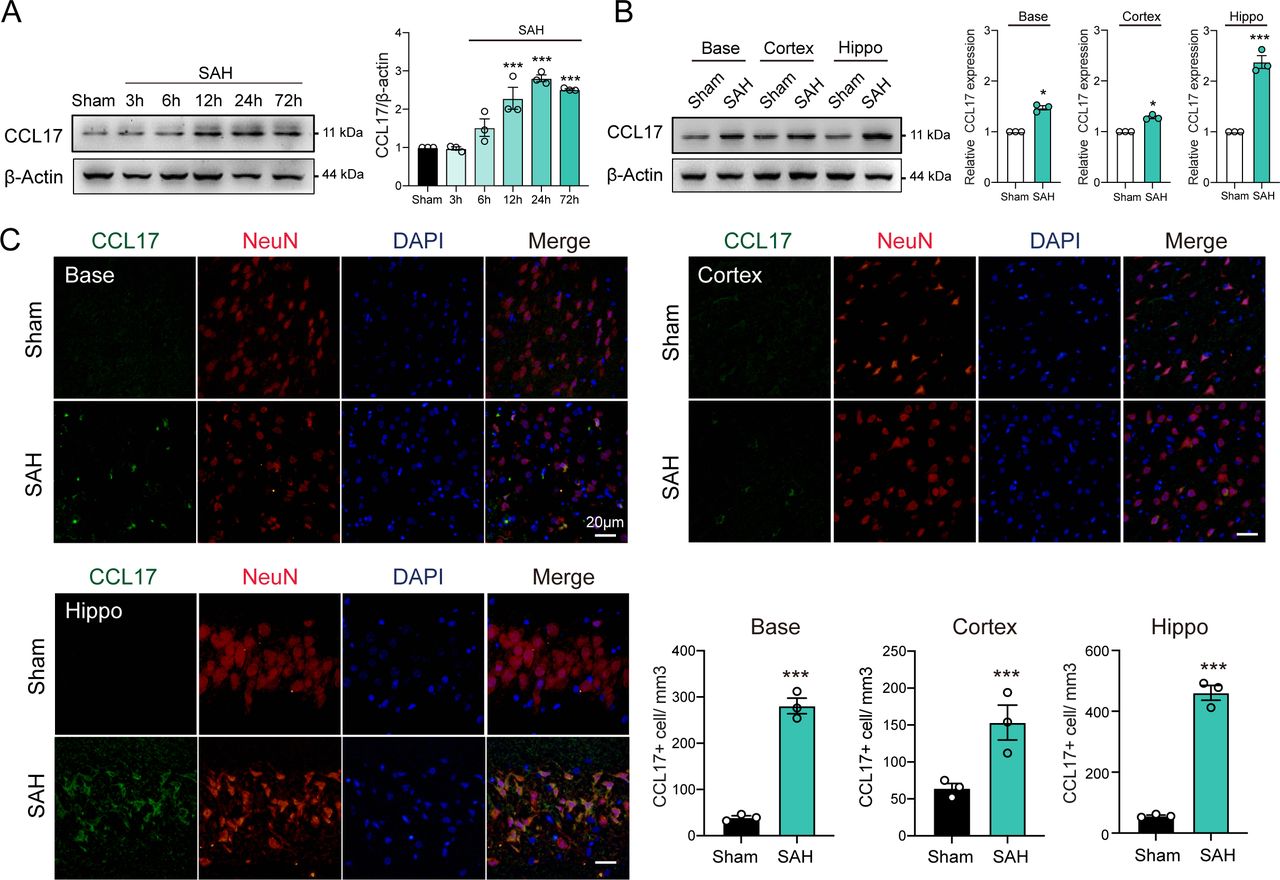
C-C motif chemokine ligand 17 (CCL17) expression was enhanced after acute subarachnoid haemorrhage (SAH) in rats. (A) Western blot analysis of CCL17 expression after SAH (n=3/group). (B) Western blot analysis of CCL17 expression at the base, cortex, and hippocampus regions at 24 hours post-SAH (n=3). (C) CCL17 (green) co-localised with NeuN (red) in basal ganglia, temporal cortex and hippocampus region in sham and 24 hours after SAH. *p < 0.05; ***p <0.001. Nuclei were stained with DAPI (blue), magnification: 200×, scale bar=20 µm, n=3. DAPI, 4',6-diamidino-2-phenylindole.
Notably, under stimulation of OxyHb, CCL17 expression in rat primary neurons was significantly increased (online supplemental figure 3A). Similarly, the CCL17 in supernatant was markedly increased (online supplemental figure 3B). These findings suggested that the expression of CCL17 was increased after haemorrhage and then secreted into the intracerebral environment.
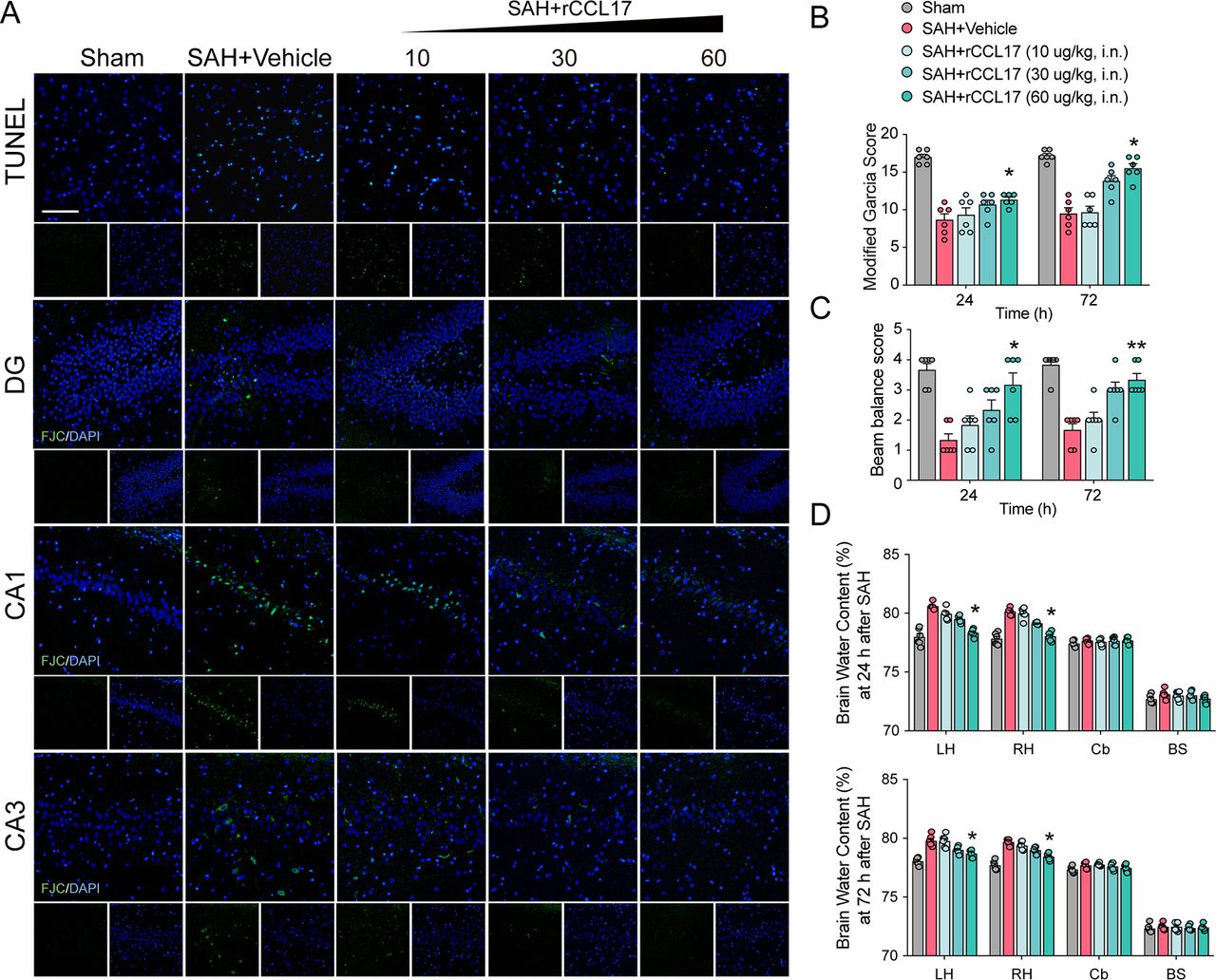
rCCL17 reduced neuronal apoptosis and BWC, but improved short-term neurobehavioural scores after SAH
Neurobehavioural scores, brain oedema, cell apoptosis and neuronal degeneration are important indicators of EBI, which is responsible for the poor prognosis of SAH.23–25 To determine the biological function of CCL17 on EBI in rat SAH model, rCCL17 was administered intranasally after SAH. There were no statistical significance in SAH scores among groups (online supplemental figure 4A). The neurological scores were markedly improved and the BWC in the left hemisphere was observably reduced with rCCL17 treatment at 24 hours and 72 hours post-SAH (figure 2B–D). Based on the above results, administration of rCCL17 (60 μg/kg) was selected for further studies.

Administration of recombinant C-C motif chemokine ligand 17 (rCCL17) reduced neuronal apoptosis and brain water content, and improved short-term neurological function after subarachnoid haemorrhage (SAH). (A) TUNEL staining in the ipsilateral cortex and FJC staining in the hippocampal area at 24 hours after SAH. Magnification: 200×, scale bar=20 µm, n=3. Modified Garcia test (B), beam balance score (C) and brain water content (D) at 24 or 72 hours after SAH revealed that rCCL17 treatment (60 μg/kg) improved short-term neurological function compared with vehicle or low dosage groups (n=6). *p < 0.05; **p < 0.01. DG, dentate gyrus; i.n., intranasal; LH, left hemisphere; RH, right hemisphere; Cb, cerebellum; BS, brain stem.
Cell apoptosis was assessed by TUNEL staining to evaluate in the ipsilateral cortex after SAH, and neuronal degeneration in the hippocampus was examined by FJC staining. Additionally, TUNEL-positive cells and FJC-positive neurons were both increased in the vehicle-treated SAH group than the sham group, which was alleviated by administration of rCCL17 (figure 2A).
The expression level and cellular localisation of CCR4 after SAH
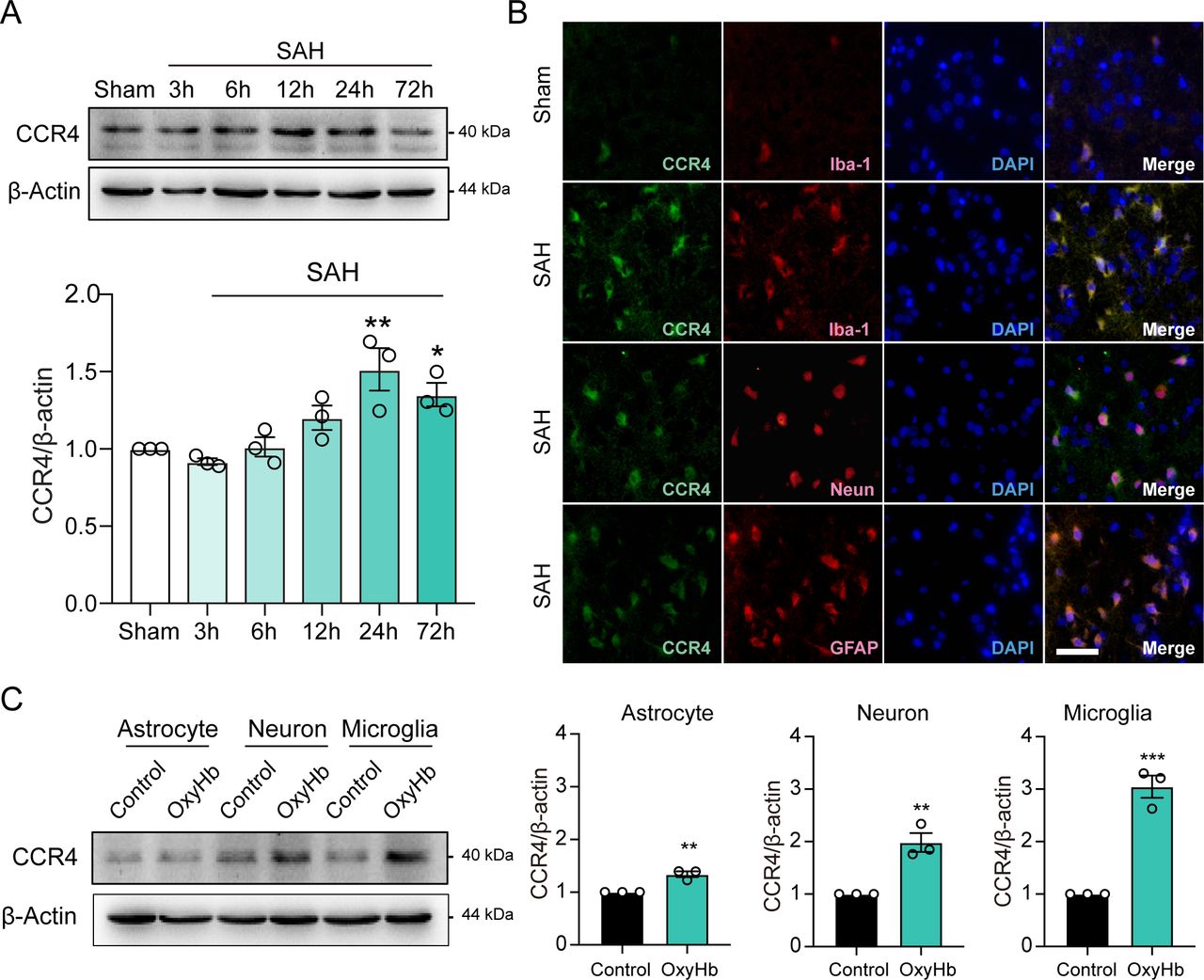
We further investigated the dynamic changes in CCR4, CCL17-specific receptor, expression in sham as well as 3 hours, 6 hours, 12 hours, 24 hours and 72 hours after SAH. Compared with the sham group, CCR4 expression was also markedly increased and peaked at 24 hours post-SAH (figure 3A). IF staining showed that CCR4 was located on microglia, neurons and astrocytes at 24 hours post-SAH (figure 3B).

Time course and co-location of endogenous C-C motif chemokine receptor 4 (CCR4) expression after subarachnoid haemorrhage (SAH). (A) Western blot analysis of CCR4 expression after SAH in rats (n=3). (B) Co-localisation of CCR4 (green) with microglia (Iba-1, red), astrocytes (GFAP, red) and neurons (NeuN, red) in the ipsilateral cortex at 24 hours after SAH in rats. Magnification: 200×, scale bar=20 µm, n=3. Nuclei were stained with DAPI (blue), scale bar=20 µm. (C) Western blot analysis of CCR4 expression in primary astrocyte, neuron and microglia cultures after treatment with haemoglobin for 24 hours (n=3/group). GFAP, glial fibrillary acidic protein.
To determine the main target cell of CCL17, we compared the level of CCR4 in various cell types after OxyHb stimulation. Interestingly, the results indicated that CCR4 expression was higher in rat primary microglia than in primary neurons and primary astrocytes in vitro (figure 3C). Above findings suggest that CCL17 may present a critical role in post-SAH microglial regulation.
Microglia exhibit an altered morphology after rCCL17 administration
To clarify the regulatory function of CCL17 on microglia in rats after SAH, we observed the morphology of microglia at 24 hours post-SAH with rCCL17 administration. As depicted in figure 4A, visual inspection of histological images suggested that the microglia of SAH rats have an altered morphology in ipsilateral cortex after rCCL17 administration. Therefore, we performed an in-depth analysis of microglial morphology in the ipsilateral cortex. The quantification of various parameters of microglial morphology in fluorescently labelled brain sections was analysed with a set of custom-written ImageJ plugins. Figure 4A depicts the representative images of individual microglia cells from each group. The respective cells were skeletonised and extracted to calculate the number of branches, end points and the process lengths. Morphological analysis demonstrated that after SAH induction, microglia present with fewer branches and end points, which were shorter in length than the sham group (figure 4B). Moreover, the M2-like microglia-associated genes (CD301, MRC1 and ARG1) were significantly decreased after SAH induction when compared with sham group, which was reversed with administration of rCCL17 in a dose-dependent manner (figure 4C). The above results revealed that microglia displayed a relatively strong increase in M2-like polarity with treatment of rCCL17.

Altered morphology and M2-like polarisation of microglia with recombinant C-C motif chemokine ligand 17 (rCCL17) treatment after subarachnoid haemorrhage (SAH) in rats. (A) Altered morphology of microglia in the ipsilateral cortex with a concentration gradient treatment of rCCL17 at 24 hours after SAH. Right panel: representative cell subjected to image analysis combined with three-dimensional representation of the determined skeleton. Magnification: 200×, scale bar=20 µm, n=3. (B) Quantification of branches, process length and end points per single cell. (C) Relative messenger RNA expression of M2-like associated genes (CD301, MRC1 and ARG1) in brain tissue post-SAH (n=4). **p < 0.01, ***p < 0.001.
rCCL17 promoted M2-like polarisation of microglia via mTORC2 signalling
To explore the function and underlying mechanisms of CCL17 on microglia polarisation, we stimulated the primary microglia with rCCL17. According to the analysis of messenger RNA (mRNA) expression of M2-like microglia-associated markers (CD301, MRC1 and ARG1), we revealed that rCCL17 can markedly promote the M2-like polarisation of microglia (p<0.05, figure 5A). Similarly, the IF also suggested that microglia tend to exert M2-like polarisation with treatment of rCCL17 (figure 5B).
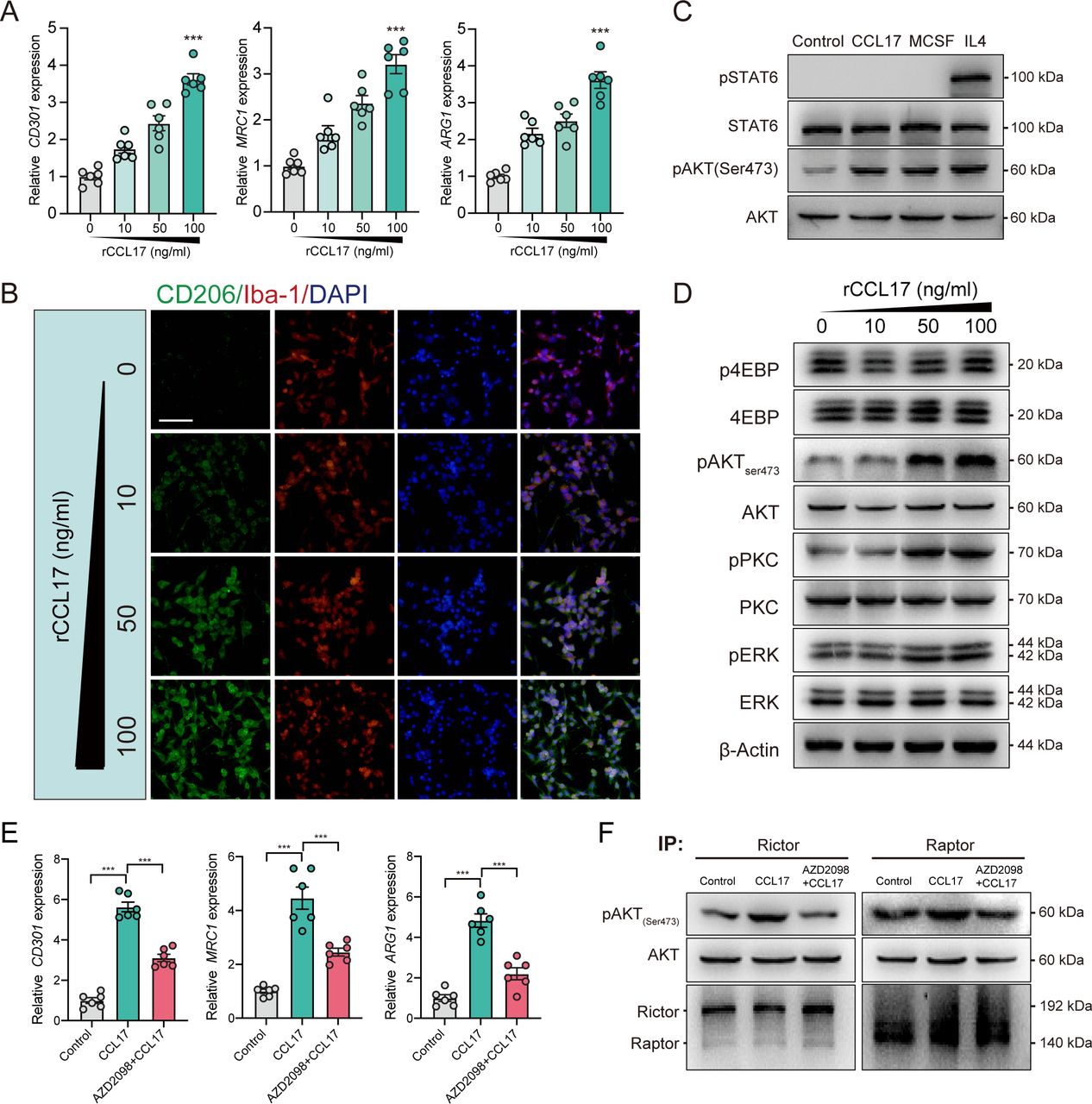
C-C motif chemokine receptor 4 (CCR4)/mammalian target of rapamycin complex 2 (mTORC2) mediates C-C motif chemokine ligand 17 (CCL17)-induced M2-like polarisation in primary microglia culture. (A) Representative M2-like microglia marker gene expression of microglia stimulated with a concentration gradient of recombinant CCL17 (rCCL17), n=6. (B) Altered morphology of microglia staining with CD206 (green) and Iba-1 (red) after stimulation with a concentration gradient of rCCL17. Magnification: 200×, scale bar=20 µm. (C) Western blot analysis showed effects of rCCL17 (100 ng/mL) treatment on STAT6 and AKT(ser473) phosphorylation in microglia, n=3/group. (D) Representative immunoblots showing effects of rCCL17 (100 ng/mL) treatment on mTORC2 downstream targets and ERK activation in microglia. (E) Relative M2-like microglia marker gene expression, n=3/group. (F) Examination of mTORC2 activity through in vitro kinase assay in rat primary microglia stimulated with rCCL17 or rCCL17 combined with AZD2098. ***p < 0.001.
It has been demonstrated that M2-like phenotype of macrophages/microglia are maintained primarily via IL4-STAT6 and MCSF-mTORC2 signalling.26 27 To investigate the mechanism of rCCL17 on M2-like microglia regulation, we first examined the influence of rCCL17 on these two pathways in primary microglia. Surprisingly, rCCL17 (100 ng/mL) did not activate IL4-induced STAT6 phosphorylation, but it greatly promoted mTORC2 activation, as shown by the increased phosphorylation of mTORC2 downstream targets, pAKT (Ser473), pPKC(Ser657) and pERK, but not mTORC1 targets, p4EBP (Thr37/46), in a dose-dependent manner (figure 5C and D).
CCL17 is the specific ligand of CCR4. Therefore, we administered the CCR4 antagonist, AZD2098, to the rCCL17-stimulated primary microglia. As shown in figure 5E, the biomarkers of M2-like polarisation were significantly increased in rCCL17-treated microglia, which was abrogated by AZD2098 (p<0.05, figure 5E). Furthermore, to directly evaluate the influence of rCCL17 on mTORC2 activity, we performed an in vitro kinase assay. The results demonstrated that rCCL17-induced phosphorylation of purified AKT at Ser473 by mTORC2 was reduced by CCR4 antagonist, whereas the mTORC1 complex did not show such significance (figure 5F). Taken together, we demonstrated that CCL17 can directly activate mTORC2 signalling through binding to CCR4 and participating in regulation of microglial functions.
rCCL17 treatment participates in neuroprotection and immune regulatory functions via CCR4/mTOCR2 axis
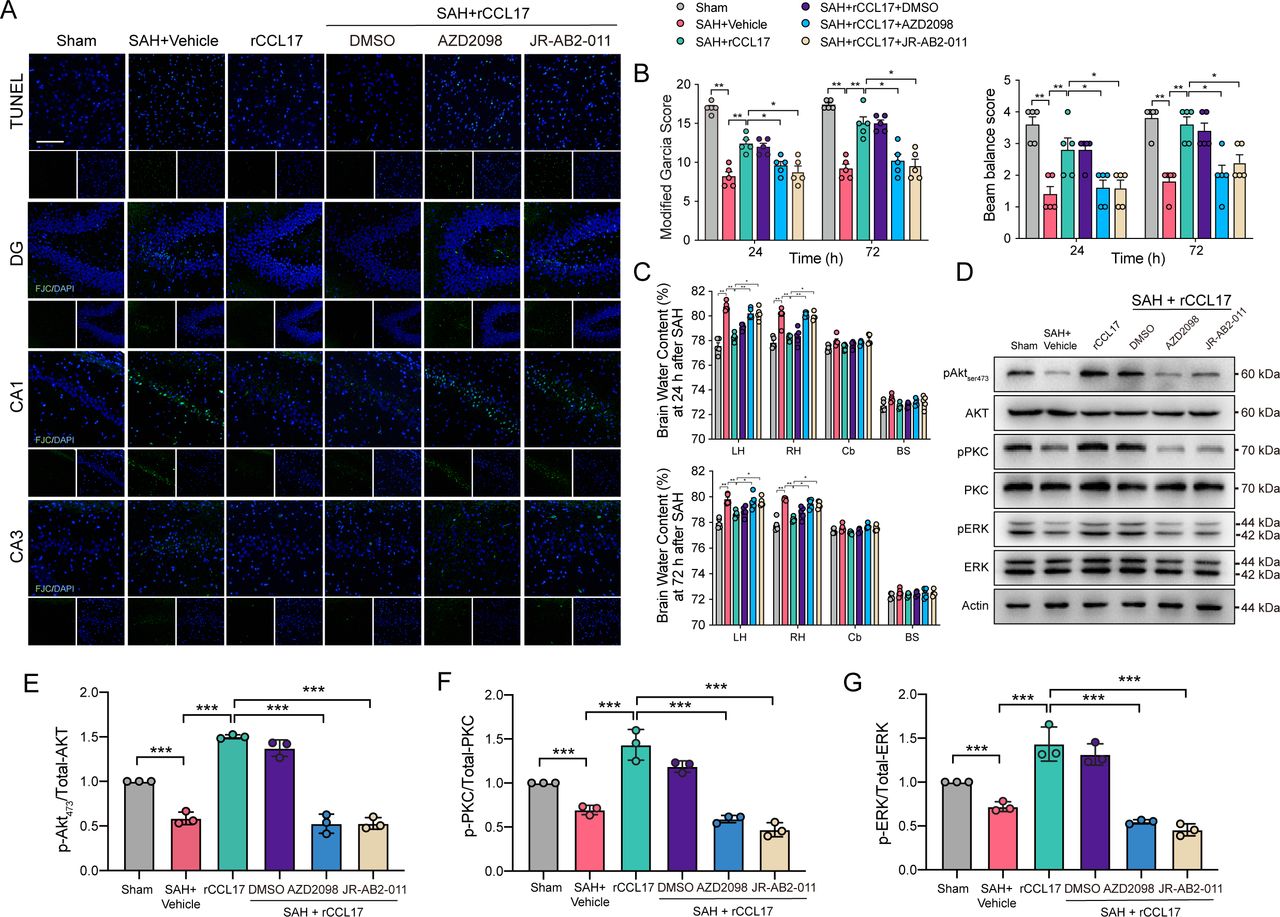
To further clarify the role of CCL17/CCR4/mTORC2 axis post-SAH, CCR4 inhibitor, AZD2098, and mTORC2 inhibitor, JR-AB2-011, were administered via intraperitoneal injection 1 hour before SAH. There were no statistical significance in SAH scores among groups (online supplemental figure 4B). TUNEL-positive cells and FJC-positive neurons were significantly decreased in the rCCL17-treated SAH group compared with the vehicle group, which was abolished in the SAH+rCCL17+AZD2098 group when compared with SAH+rCCL17 and SAH+rCCL17+DMSO group (figure 6A). Additionally, pretreatment with the CCR4 antagonist or the mTORC2 selective inhibitor abrogated the neurobehavioural benefits of rCCL17 in EBI post-SAH (figure 6B and C). Western blot analysis revealed that after rCCL17 administration, the mTORC2 signalling pathway was activated, compared with both the SAH+vehicle and sham group, which was markedly inhibited after treatment with either AZD2098 or JR-AB2-011 (figure 6D-G).

The effect of recombinant C-C motif chemokine ligand 17 (rCCL17)/C-C motif chemokine receptor 4 (CCR4)/mammalian target of rapamycin complex 2 (mTORC2) axis was reversed by administration of CCR4 inhibitor and mTORC2 inhibitor after subarachnoid haemorrhage (SAH) in rats. (A) TUNEL staining in the ipsilateral cortex and FJC staining in the hippocampal area at 24 hours after SAH. Magnification: 200×, scale bar=20 µm, n=3. Modified Garcia test (B), beam balance score (C) and brain water content at 24 or 72 hours post-SAH showed that the activation of CCR4/mTORC2 axis with rCCL17 treatment was abolished by CCR4 inhibitor, AZD2098 and mTORC2 inhibitor, JR-AB2-011, n=5. (D) Western blot analysis of phosphorylation of AKT473 (E), PKC (F) and ERK (G) were performed. *p < 0.05, **p < 0.01, ***p < 0.001.
Due to the regulatory function of CCL17 on microglia, we also analysed the altered morphology and M2-like polarisation of microglia in the ipsilateral cortex. Visual inspection of the Iba-1 staining sections indicated that microglia possessed fewer branches and end points, which were also shorter in length after treatment with AZD2098 or JR-AB2-011 (figure 7A and B). Furthermore, the biomarkers of M2-like polarisation was also markedly decreased after administration with both inhibitors (p<0.05, figure 7C). Together, the above results illustrated the neuroprotective role and immune regulatory functions of the CCL17/CCR4/mTORC2 signalling pathway post-SAH.

Recombinant C-C motif chemokine ligand 17 (rCCL17) induced M2-like polarisation of microglia was reversed by inhibition of C-C motif chemokine receptor 4 (CCR4)/mammalian target of rapamycin complex 2 (mTORC2) axis after subarachnoid haemorrhage (SAH) in rats. (A) Altered morphology of microglia in the ipsilateral cortex from Sham or SAH rats model with treatment of rCCL17 and administration of CCR4 inhibitor, AZD2098 and mTORC2 inhibitor, JR-AB2-011, at 24 hours post-SAH. Right panel: representative cell subjected to image analysis combined with three-dimensional representation of the determined skeleton. Magnification: 200×, scale bar=20 µm, n=3. (B) Total number of branches, process length and end points per single cell were calculated. (C) Relative messenger RNA expression of M2-like associated genes (CD301, MRC1 and ARG1) in brain tissue at 24 hours post-SAH. All t-tests were two-tailed, n=4/group. *p < 0.05, **p < 0.01, ***p < 0.001.
CCL17-regulated microglia promoted neuronal apoptosis and neurobehavioural deficits via CCR4/mTORC2 signalling
According to the above results, we demonstrated that rCCL17 treatment alleviated EBI via CCR4/mTORC2 signalling. However, the application of targeted protein antagonists could not exclude off-target effects, nor could the targeting of the specific cells because of the complex network in the brain. Thus, due to the significant rise of CCR4 in microglia after SAH and its immune regulatory functions on microglia, we hypothesised as to whether rCCL17-regulated microglia could mediate the benefits of rCCL17 treatment. Rictor is a component of the mTORC2 complex and mediates the activity of mTORC2.28 Therefore, we pre-injected AAV-CD68-Control and AAV-CD68-shRictor before SAH induction to target and knockdown the expression of Rictor and inhibit the function of the mTORC2 complex in microglia/macrophages in the brain. There were no statistic significance in SAH scores among groups (online supplemental figure 4C).
As shown in figure 8, microglia possessed significantly fewer branches and end points, which were shorter in length after knockdown of Rictor expression in microglia in ipsilateral cortex (figure 8A and B). Furthermore, the M2-like polarisation biomarkers were also markedly decreased after intracerebroventricular injection of adeno-associated virus (figure 8C). Similarly, the TUNEL-positive cells and FJC-positive neurons were both decreased by rCCL17 treatment in the AAV-CD68-Control-treated group, whereas the AAV-CD68-shRictor-treated group did not demonstrate such tendency (figure 9A). Moreover, consistent with the above results, in the AAV-CD68-Control-treated group, the neurobehavioural score was improved and the BWC in the left hemisphere was significantly augmented with rCCL17 treatment at 24 hours and 72 hours compared with the SAH+vehicle group. However, the beneficial effect of rCCL17 treatment was abolished in the AAV-CD68-sRictor-treated group (figure 9B-H). These results demonstrated that by targeting and inhibiting the CCR4/mTORC2 axis in microglia, the neuroprotective effects of CCL17 can be abrogated.
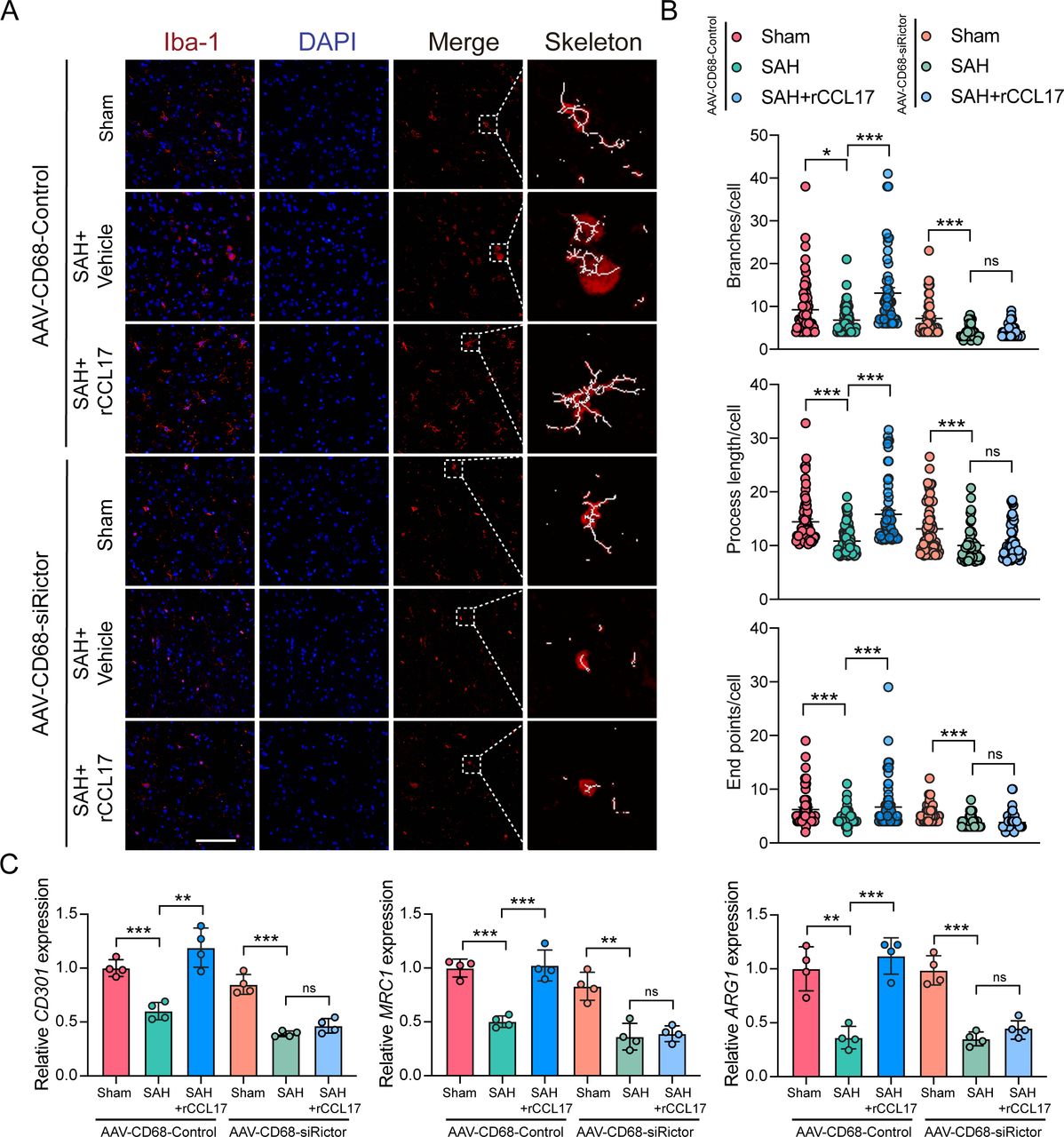
Recombinant C-C motif chemokine ligand 17 (rCCL17) induced M2-like polarisation of microglia was reversed by the inhibition of mammalian target of rapamycin complex 2 (mTORC2) signal pathway. Sprague-Dawley rats were intracerebroventricularly pretreated with AAV-CD68-Control or AAV-CD68-shRictor before surgery to target and knockdown the Rictor expression in microglia. (A) Representative images of microglia in the ipsilateral cortex from Sham or subarachnoid haemorrhage (SAH) rats model with treatment of rCCL17 at 24 hours post-SAH. Right panel: representative cell subjected to image analysis combined with three-dimensional representation of the determined skeleton. Magnification: 200×, scale bar=20 µm, n=3. (B) Total number of branches, process length and end points per single cell were calculated. (C) Relative messenger RNA expression of M2-like associated genes (CD301, MRC1 and ARG1) in brain tissue at 24 hours post-SAH, n=4/group. ns, not significant. *p < 0.05, **p < 0.01, ***p < 0.001.

Activation of C-C motif chemokine receptor 4 (CCR4)/mammalian target of rapamycin complex 2 (mTORC2) axis in microglia mediated the neuroprotective function of recombinant C-C motif chemokine ligand 17 (rCCL17). Sprague-Dawley rats were intracerebroventricularly pretreated with AAV-CD68-Control or AAV-CD68-shRictor before surgery to target and knockdown the Rictor expression in microglia. (A) Representative image of the TUNEL in the ipsilateral cortex and FJC staining in the hippocampal area at 24 hours postsubarachnoid haemorrhage (SAH). Magnification: 200×, scale bar=20 µm, n=3. Modified Garcia test (B), beam balance score (C) and brain water content at 24 or 72 hours after SAH showed that the neuroprotective effect of rCCL17 was mediated through the activation of CCR4/mTORC2 axis in microglia, n=5/group. (D) Western blot analysis of the quantification of Rictor (E), phosphorylation of AKT473 (F), PKC (G) and ERK (H) were performed. ns, not significant. *p < 0.05, **p < 0.01, ***p < 0.001.
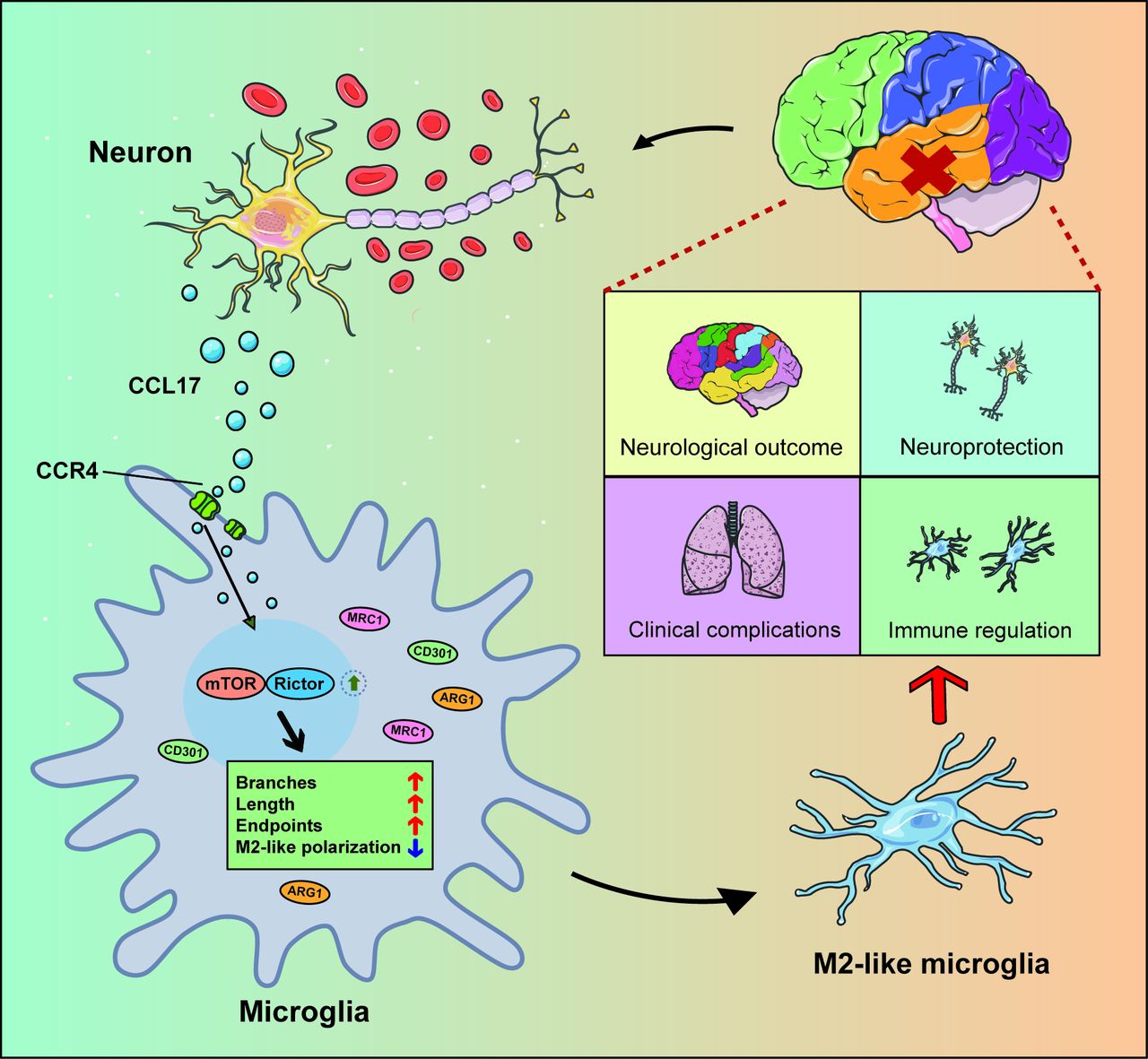
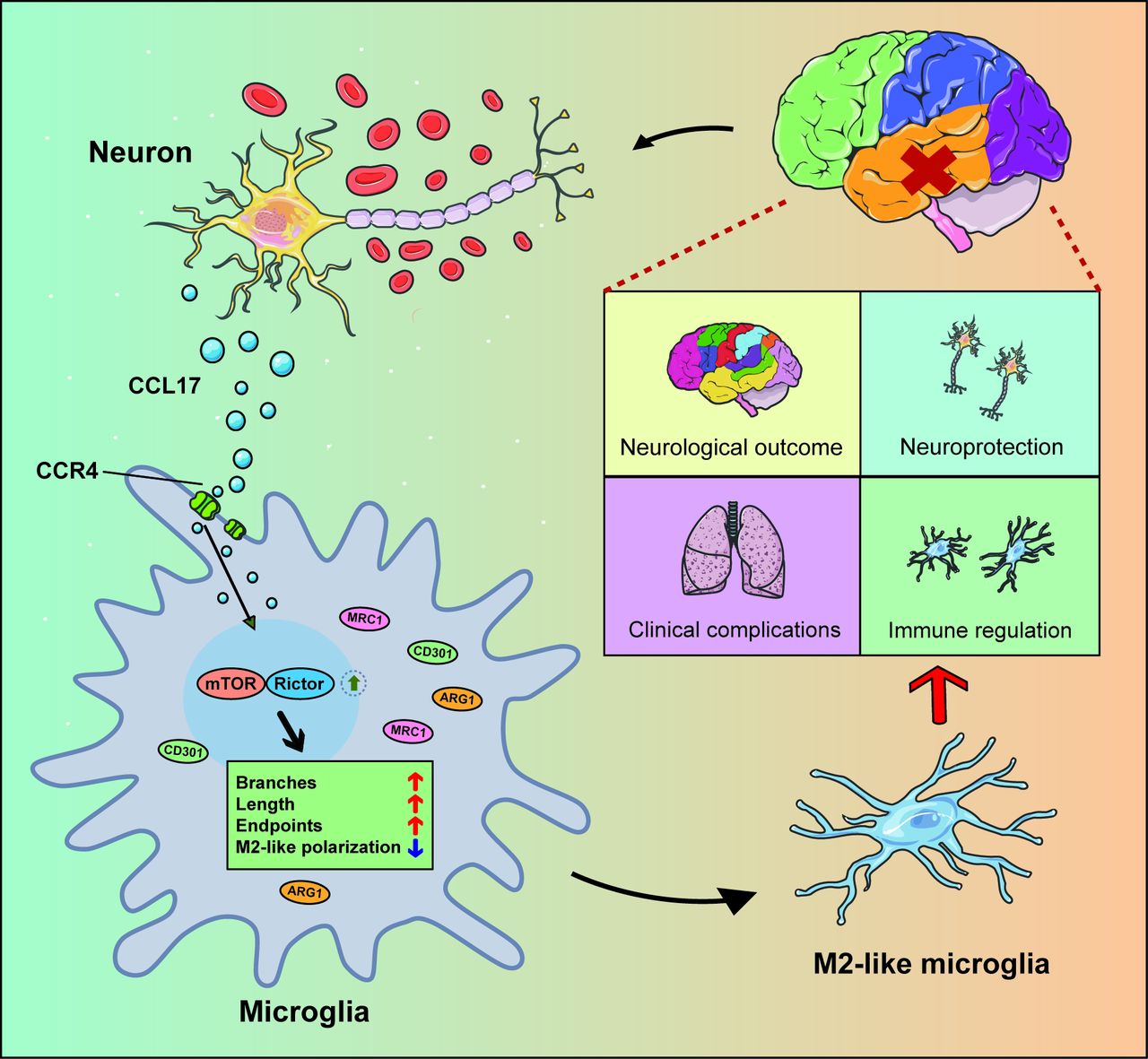
In summary, these data (figure 10) illustrated that CCL17 secreted from neurons in the setting of haemorrhage can promote the M2-like polarisation of microglia to then exert neuroprotective functions post-SAH.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic model showing the mechanism of C-C motif chemokine ligand 17 (CCL17)-mediated crosstalk between neuron and microglia attenuate neurological injury after subarachnoid haemorrhage (SAH). Overproduction of CCL17 presents an essential role in neuroprotection and immune regulation. Following SAH onset, CCL17 secreted by neuron promotes microglia towards M2-like phenotype polarisation via the C-C motif chemokine receptor 4 (CCR4)/mammalian target of rapamycin complex 2 (mTORC2) axis. In return, the M2-like microglia present neuroprotective functions and attenuate neurological injury.
Discussion
The highlights of this study are listed as follows: (a) rCCL17 treatment improved EBI after SAH and promotes M2-like polarisation of microglia through the CCR4/mTORC2 signal pathway in rats; (b) inhibition of CCR4 in microglia will abolish the neuroprotective effects of rCCL17. In this study, we demonstrated that administration of rCCL17 can alleviate nervous system lesions, at least in part, through the CCR4/mTORC2 signalling pathway, and improved outcomes after SAH.
CCL17 has recently received substantial attention due to its role in a wide range of physiological processes. In a previous study, homozygous CCL17/EGFP knockout mice (CCL17E/E) revealed the location of Ccl17 expression in the murine brain. Notably, CCL17+ cells were clearly observed in the region of hippocampus and the result of co-staining with neuronal marker NeuN revealed that CCL17 is located in neurons within the CA1 region.20 Consistently, according to the in vitro experiments of rat primary neurons, CCL17 expression in neurons was significantly increased and secreted into the microenvironment. The evidence above implied that neurons may be the partial source of secreted CCL17 after SAH. In addition, we demonstrated that rCCL17 administration notably alleviated the SAH-induced neurological deficits and brain oedema.
Accumulative evidence have illustrated that activated microglia/macrophages can induce neuronal apoptosis in the early period following SAH.29 30 Microglial polarisation to selectively activate M2-like phenotypes facilitated tissue repair on acute brain injury and spinal cord injury.10–12 31 Additionally, it has also been demonstrated that microglia presented a crucial role in several complications following SAH, including cerebral vasospasm32 33 and symptomatic epilepsy.34 Recent studies have revealed that anti-inflammatory M2 microglia exerted neuroprotective functions and alleviated brain oedema. In a previous study, IL-4-induced M2 microglia markedly alleviated neurological damage (including neuronal death and brain oedema) after ICH via the JAK1/STAT6 pathway.14 It has also been demonstrated that M2 microglia-derived exosomes promoted neuronal survival and attenuated ischaemic brain injury.13 Moreover, in terms of SAH, accumulative evidence have revealed that inducing shift towards M2 phenotype or inhibiting M1-like polarisation could ameliorate EBI and improve neurological functions in SAH models.35 36 Thus, targeting M2 microglia may present a promising strategy to ameliorate brain injury after SAH. We observed that the M2-like microglia-associated genes were significantly decreased after SAH induction, which was reversed with administration of rCCL17.
To study the underlying mechanism of rCCL17 on M2-like microglia regulation, we found that rCCL17 greatly promoted activation of mTORC2 signalling. Combined with the direct in vitro kinase assay, we examined the direct influence of rCCL17 on mTORC2 activity. In line with this, we observed that the CCL17 functions of promoting EBI and M2-like polarisation were abolished with administration of CCR4 inhibitor or mTORC2 inhibitor. Application of antagonists could not exclude off-target effects, nor could targeting specific cells. Therefore, we specifically downregulated the expression of Rictor in microglia to suppress the mTORC2 complex via AAV-CD68-shRictor. Consistently, targeting microglia via AAV can also abrogate the promoting function of CCL17. Taken together, we demonstrated that CCL17 can directly activate mTORC2 signalling through binding to CCR4 and regulating microglial functions.
However, there are several shortcomings in this study: (1) CCR4 was localised in various cells. We only clarified the role of CCL17 in regulation of microglia-mediated neuroinflammation post-SAH; (2) although this article indicated that binding to CCR4 changes the activity of mTORC2 complex and regulates M2-like polarisation, the mechanism remains to be unveiled, and further experimentation is warranted; (3) the expression and secretion levels of CCL17 are not measured in other types of neural cells. Therefore, neuron-derived CCL17 may be the partial source of CCL17 after SAH. The dominant source of endogenous CCL17 following SAH requires further investigation and more in-depth exploration in subsequent studies.
Conclusions
In this study, we confirmed that CCL17 attenuated neurological deficits after SAH via activation of the CCR4/mTORC2 axis in microglia. These findings offer novel insight into the critical role of CCL17 in M2-like polarisation of microglia, which provide a novel approach to the clinical strategy targeting microglial polarisation in early SAH management.
Data availability statement
Data are available on reasonable request. All raw data used in this manuscript are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
All animal experiments were performed according to the Institutional Animal Care and Use Committee (IACUC). The procedures were conducted according to the National Institutes of Health’s Guide for the Care and the Use of Laboratory Animals and the Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines.
References
Supplementary material
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
AZ, YL and HX contributed equally.
Contributors SC, JZ and YF are the principal investigators and designed the study. AZ, YL, HX and ZZ contributed to the study design, experiment implementation and manuscript draft. XW, LY, CL and CZ summarised and analysed the experimental data and made suggestions to improve the study. JJ and CF revised the manuscript and polished the language. The author(s) read and approved the final manuscript. AZ is responsible for the overall content as the guarantor.
Funding This work was supported by the National Natural Science Foundation of China (81870916 and 81971107) and the Fundamental Research Funds for the Central Universities, China (2019QNA7038).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.