Article Text

Abstract
Cerebral cavernous malformations (CCMs) are vascular lesions characterised by enlarged and irregular structure of small blood vessels in the brain, which can result in increased risk of stroke, focal neurological defects and seizures. Three different genes, CCM1/Krev/Rap1 Interacting Trapped 1, CCM2/MGC4607 and CCM3/PDCD10, are associated with the CCMs’ progression, and mutations in one of three CCM genes cause CCM disease. These three CCM proteins have similar function in maintaining the normal structure of small blood vessels. However, CCM3 mutation results in a more severe form of the disease which may suggest that CCM3 has unique biological function in the vasculature. The current review focuses on the signalling pathways mediated by CCM3 in regulating endothelial cell junction, proliferation, migration and permeability. These findings may offer potential therapeutic strategies for the treatment of CCMs.
- CCM3
- PDCD10
- cell junction
- angiogenesis
- GCKⅢ
- EndMT
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Cerebral vascular malformations affect 0.1%–4% of the general population and fall into four categories: arteriovenous malformations, venous malformations, capillary telangiectases and cerebral cavernous malformations (CCMs). CCMs consist of clusters of enlarged endothelial channels (‘caverns’) that are arranged back-to-back to form densely packed sinusoids with little or no intervening brain parenchyma.1–5 These lesions lack smooth muscle/pericyte and elastic tissue, and lack sub-endothelial support and an intact basal lamina, so the vessel walls are thin and prone to leakage. Ultra-structural analysis has revealed decreased numbers of pericytes, endothelial detachment from the basal lamina and ruptures in the luminal endothelium probably due to reduced/damaged intercellular junctions.6 CCMs are primarily found within the neurovasculature of the central nervous system (CNS, ie, brain, spinal cord, retina), where they result in increased risk for stroke, seizures and focal neurological deficits.3–5 Currently, the only treatment for CCM is surgical resection. CCMs may be familial or sporadic. Familial cases are caused by mutations in one of three CCM genes: CCM1 (also known as Krev/Rap1 Interacting Trapped 1—KRIT1),7 CCM2 (also known as malcavernin or osmosensing scaffold for mitogen-activated protein kinase kinase-3-osm)8 and CCM3 (also known as programmed cell death 10-PDCD10).9 These three CCM proteins can be found in the same complex within the cell and recent studies suggest that all CCM proteins regulate EndMT (endothelial-to-mesenchymal transition), which contributes to the onset and progression of CCM.10–15 However, CCM3 might also act separately from CCM1 and CCM2, as its mutation in humans often results in a more severe form of the disease,16 17 and CCM3 knockout mice show severe phenotypes with yet-to-be defined mechanisms.10–14
Bergametti et al first identified PDCD10 as the third CCM gene, which is located on 3q25.2–27.9 After the relationship between PDCD10 and CCMs was elucidated, a variety of biological functions of this protein related to CCMs have been reported. This review focuses on the role of CCM3 in regulating cell junction, maintaining normal structure and function of vascular endothelial cells, and angiogenesis, which all can be involved in the progression of CCMs.
CCM3 and the cerebrovascular disease
The role of CCM3 in cell junction
The normal cell junction of endothelial cells is important to maintain the normal structure and function of blood vessels, which are often disrupted in CCM lesions. Cell-cell junctions especially tight junctions are essential to prevent blood-borne compounds from leaking to brain parenchyma, which further leads to inflammatory responses, endothelial injury and lesion progression.18 19 Stamatovic et al 20 have found that CCM3 regulates brain endothelial barrier integrity and tight junction complex organisation. Loss of CCM3 activates extracellular signal-regulated kinase (ERK1/2); activated ERK1/2 induces Ser phosphorylation (pS405) of cortactin (a cortical actin ring protein), which leads to cortactin degradation. Increased cortactin degradation is associated with loss of the cortical actin ring and ZO-1: actin interactions, which are essential for providing physical support and anchoring of tight junction proteins. The reduced anchoring of ZO-1 to the actin cytoskeleton impacts organisation of tight junction proteins, and eventually disrupts the tight junction complex.
CCM3 also may affect tight junction by indirectly interaction with CCM1, as CCM1 has been proved to maintain Rap-1 mediated stabilisation of endothelial junction and VE-Cadherin mediated adherens junctions.21 22 CCM3 and germinal-centre kinase III (GCKIII) family associated with striatin also interact with the CDC42 binding kinase myotonic dystrophy kinase-related CDC42-binding kinase, which promotes circumferential actin bundles essential for junction formation.23
The role of CCM3 in angiogenesis
Angiogenesis is a process that involves endothelial cells proliferation, migration and morphology remodelling. He et al 24 have shown that CCM3 knockdown in human umbilical vein endothelial cells significantly reduces endothelial cell proliferation and induced cell apoptosis, and also inhibits vascular endothelial growth factor (VEGF-induced endothelial cell) cord formation. CCM3 specifically associates with VEGFR2 and was required for stabilisation of VEGFR2, thereby maintaining the VEGF signalling pathway which is essential for angiogenesis.
CCM3 can be involved in different signalling pathways as an anchor protein which can bind to different target proteins. The best-characterised interaction between CCM3 and its target proteins lies in the dimerisation-domain-mediated interaction with the GCKIII group of kinases, MST4/MASK, STK24/MST3 and STK25/YSK1/SOK1.25
Zhang et al 26 have first proved that CCM3 associates with STK24 and regulates exocytosis in neutrophils. STK24 binds to UNC13D C2B domain and prevent UNC13D from binding to lipids, a step important for vesicle docking. CCM3 can stabilise the STK24 protein, and there is a degranulation phenotype in neutrophils during loss of either CCM3 or STK24. Then Jenny Zhou et al 27 has subsequently uncovered that CCM3 regulates exocytosis in endothelial cells. CCM3 can inhibit UNC13-mediated intracellular molecules exocytosis by combining with UNC13B and STK24. Thus, loss of CCM3 increases ANGPT-2 release from Weibel-Palade bodies in brain endothelial cells. Increased ANGPT-2 secretion to the extracellular space disrupts the association between endothelial cells and pericytes, leading to enhanced endothelial cell spouting, and lumen formation followed by CCM lesion formation.
Zheng et al 28 that loss of CCM3 or STK24 also results in actin stress fibre formation and elevated RhoA activation in endothelial cells, so ANGPT-2 induced excessive sprouting and lumen formation along with weakened cell adhesion and increased permeability result in an abnormal and disrupted blood vessel.
Zhou et al 29 have reported that CCM complex associated with GCKIII kinases regulates MEKK3 pathway in endocardial cells. Endocardial deletion of CCM1/CCM2/CCM3 activates MEKK3 and the downstream MEK4 and ERK5. Activated ERK5 could be transported into the nuclei and upregulate the transcription factor KLF2/4 and proteases a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS4/5). Zhou et al 29 also demonstrate that MEKK3-KLF2/4 signalling is critical for the CCM progression.
Other mechanisms about CCM3 regulates CCM progression
EndMT is characterised by the acquisition of mesenchymal and stem-cell-like features of endothelium.30 31 The progression of EndMT leads to disrupted cell junction organisation, loss of cell polarity, increased cell proliferation and migration.32 Maddaluno et al 13 have shown that EndMT exists in CCM1 ECKO and CCM3 ECKO mice with disorganised VE-Cadherin and significantly up-regulated N-Cadherin, and this progress is mediated by the upregulation of endogenous BMP6, which in turn activates the TGF-β (transforming growth factor-β) and BMP (bone morphogenetic protein) signalling pathway. Inhibiting TGF-β or BMP pathway prevents EndMT and reduces CCM lesion in mice. Autophagy is also related to CCM formation and EndMT.33
Louvi et al 34 have found that CCM3 deletion in neural cells results in a vascular phenotype that resemble human CCMs, which suggests CCM3 may affect CCM progression through cell death pathway. Fidalgo et al 35 have also found that CCM3-GCKIII kinase (MST4) mediates ezrin/radixin/moesin phosphorylation and cell survival after reactive oxygen species (ROS) stimulating. Interestingly, ROS can be induced by CCM3 deletion in endothelial cells.33 All of these studies suggest ROS may be a factor implicated in loss of CCM3-dependent CCMs progression.
CCM3 may play an important role in other biological phenomenon besides CCM lesion. Guerrero et al 36 have reported that lack of CCM3 impairs the senescence response of cells, which is related to the inability of CCM3-deficient cells to induce the C/EBPβ transcription factor. CCM3-deficient cells also have a defect in autophagy which may suggest an evidence of immortal cells, and one study reveals a relationship between CCM3 mutation and high risk of tumour.37
Progression in CCM therapy
The only treatment for CCM disease is surgical resection so far, although there is high risk for cerebral operation. To date, no medical therapy has been approved. Based on the researches of molecular mechanism which regulate CCM progression, some drugs which affect intracellular signalling pathway can be effective in animal trials. Administration of fasudil, a Rho-kinase inhibitor, results in attenuated CCM lesion in mice with CCM1 mutations.33 Statin inhibits HMG-CoA reductase, which reduces RhoA-dependent small GTPase activation, and presents a symptomatic improvement in mouse model,38 but administration of statin is associated with increased risk of intracerebral haemorrhage,39 and hence more researches should be done before the application of statin in CCM therapy. ANGPT2-neutralising antibody significantly reduces CCM lesion formation in CCM3 ECKO mice.27 TLR4 (Toll-like receptor 4) antagonists and alteration of microbiome can affect CCM formation in mice.40 Many advances have been made, but further studies are still needed to uncover novel mechanisms regulating CCMs and develop effective drugs preventing the progression of CCMs.
Conclusion
As an anchor protein, CCM3 binds to different type of proteins; this enables CCM3 take part in different intracellular signalling which affect cell junction, angiogenesis, apoptosis and stress responses (figure 1). Some controversial results may exist related to specific CCM3-mediated pathways observed in isolated endothelial cells. The current prevailing view is that the primary defects in CCM are endothelial cells-intrinsic in humans, and this has been confirmed by endothelial cell-specific gene deletion in mouse models. We have to consider that CCM lesion is a comprehensive result of all different pathways in the neurovascular unit, which consists of several cell types, including endothelial cells, pericytes, astrocytes and neuronal cells. Despite the mounting knowledge about the role of CCM3 in the progression of CCMs, several fundamental questions need to be addressed in the CCM field. Why are CCM lesions primarily confined to brain vasculature despite CCM proteins are ubiquitously expressed in all tissues? The unique feature of the brain vasculature is the blood brain barrier (BBB) formed by the brain neurovascular unit. It is unknown whether or not the unique BBB structure and the interactions of endothelial cells with neural cells and pericytes play a critical role in CCM lesion development. We don’t know the effect of CCM3 on translational and post-translational modifications of those tight junction proteins, and how CCM3 regulates β1 integrin signalling. Recent study uncovered a relationship between innate immune/microbiome and CCM lesion in mice with CCM1/2 deficiency. Whether or not CCM3 is linked to the innate immune pathway is unclear yet. We should notice that there may be other CCM-related genes, as mutations in CCM1/CCM2/CCM3 do not cover all the familiar cases. There is no doubt that further studies are necessary to better understand the mechanism and pathogenesis of CCMs and find a non-invasive therapy for CCM disease.
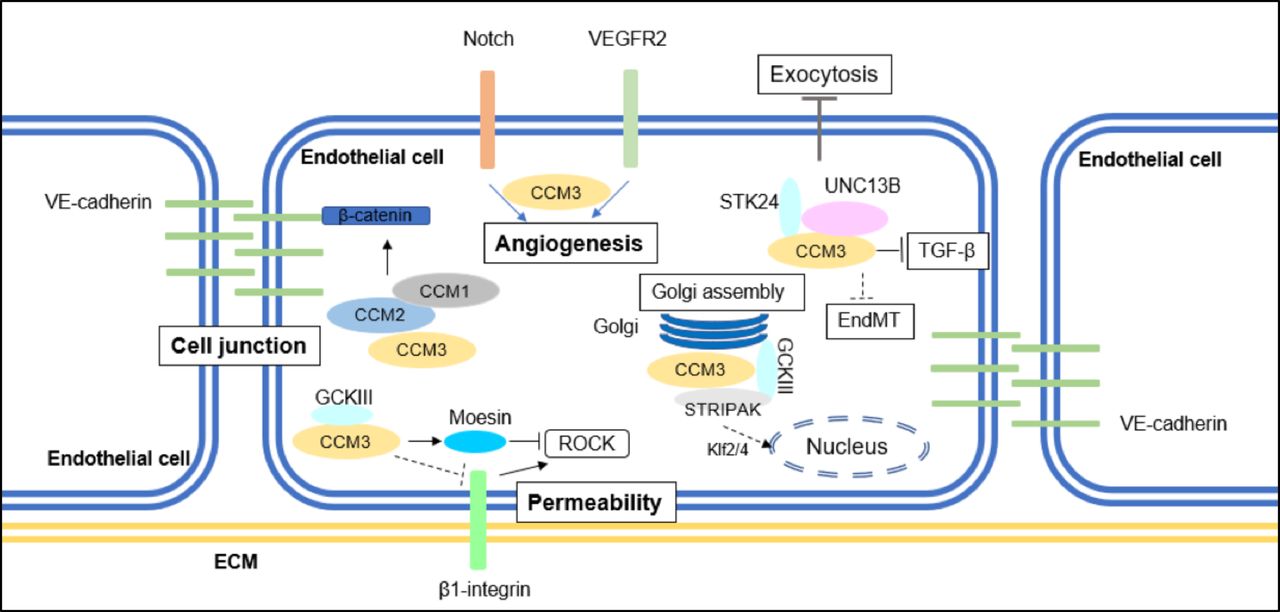
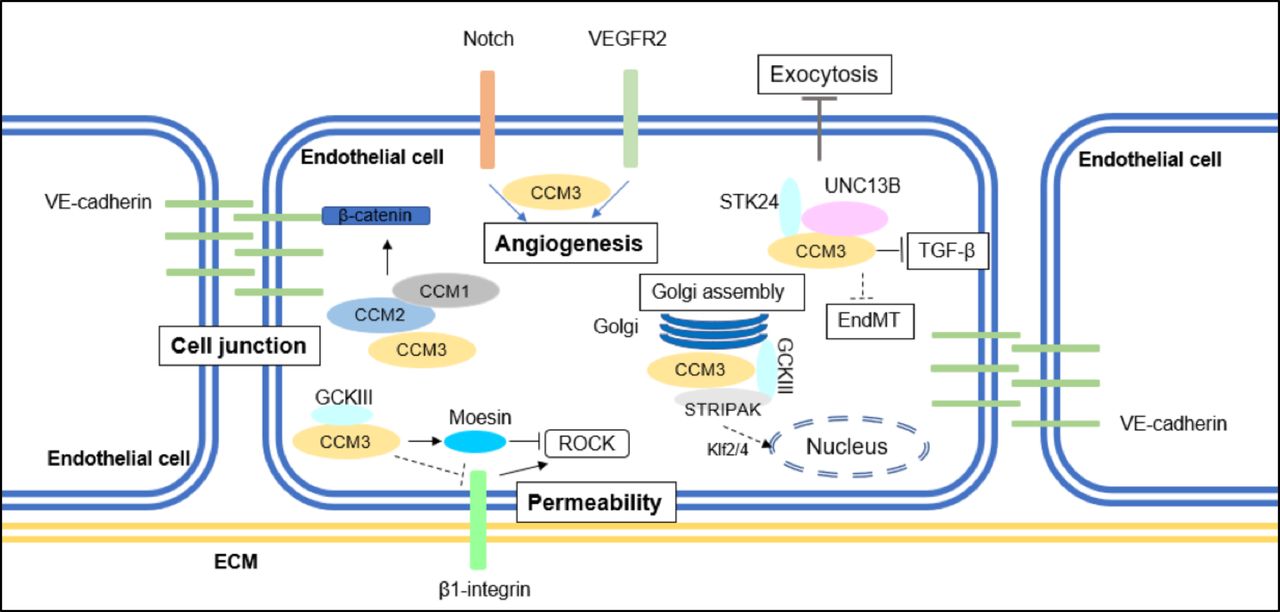{kind=link}
A summary of CCM3-mediated signalling pathways implicated in CCM formation. CCM3 maintains cell junction by inhibiting ERK1/2 phosphorylation; CCM3 inhibits stress fibre migration and endothelial permeability by inhibiting RhoA signalling; CCM3 regulates angiogenesis with or without binding to GCKIII kinases. CCM3 also mediates EndMT transition, autophagy and ROS stimulation. CCM, cerebral cavernous malformation; EndMT, endothelial-to-mesenchymal transition; GCKIII, germinal-centre kinase III; VE, vascular endothelial.
References
Footnotes
Contributors KW wrote the article, HJZ and WM provided guidance and modifications.
Funding This work was supported by National Key Research and Development Program of China (2016YFC1300600), National Natural Science Foundation of China (No. U161219), Science Grant of Guangzhou (2016040220131), and Science grants of Guangdong (No. 2015B020225002). This work was partly supported by NIH grants R01 HL136507, R01HL109420 and HL115148.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement No additional data are available.
Correction notice This article has been corrected since it was published. Dr. Min Wang’s name was incorrectly transposed.
Patient consent for publication Not required.