Article Text

Abstract
As the resident immune cells in the central nervous system, microglia have long been hypothesised to promote neuroinflammation and exacerbate neurotoxicity. However, this traditional view has undergone recent revision as evidence has accumulated that microglia exert beneficial and detrimental effects depending on activation status, polarisation phenotype and cellular context. A variety of neurotransmitter receptors are expressed on microglia and help mediate the bidirectional communication between neurons and microglia. Here we review data supporting the importance of neurotransmitter receptors on microglia, with a special emphasis on glutamate, γ-aminobutyric acid (GABA), norepinephrine, cannabinoid and acetylcholine receptors. We summarise evidence favouring a significant role for neurotransmitter receptors in modulating microglial activation, phagocytic clearance and phenotypic polarisation. Elucidating the effects of neurotransmitter receptors on microglia and dissecting the underlying mechanisms may help accelerate the discovery of novel drugs that tap the therapeutic potential of microglia.
- microglia
- neurotransmitter
- glutamate
- Noradrenaline
- Cannabinoid
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Microglia, the dynamic, motile phagocytes of the central nervous system, were traditionally viewed as proinflammatory cells that promote neuronal toxicity and death. However, this view failed to account for the dualistic roles that microglia play—roles that are profoundly shaped by existing physiological or pathological cellular conditions. Recent studies suggest that microglia are a double-edged sword, exerting toxic and beneficial roles depending on their polarisation phenotype, activation status and the cellular context.1 In addition to the previous overly simplistic view of microglia, neurons were often portrayed as passive victims of microglial activation. However, this view has also undergone revision because communication between these two cell types actually flows in both directions.2 First, it has been established that the quiescent state of microglia in the healthy brain is controlled, at least in part, by neuronal factors, such as CD200, as indicated from elevated microglial activation induced by knockout of neuronal CD200.3 Second, neuron-specific injury is known to activate nearby microglia that are associated with the damaged neurons. For example, nerve injury specifically in the peripheral nervous system activates those microglia located at the innervation site of the damaged nerve in the central nervous system.4 These observations support the view that neurons communicate their health status to the appropriate microglia. Presumably, the microglial recipients of neuron-derived information in turn influence neuronal viability in positive and negative feedback loops, consistent with their dualistic roles in health and disease. However, even though microglia promote neuronal loss in some models, this glial behaviour probably originally evolved for the efficient removal of irreparably injured, highly dysfunctional cells to prevent further damage to the organism as a whole. Indeed, the bidirectional nature of microglial–neuronal communication might be the foundation for healthy crosstalk between neurons and glia before a final decision about neuronal life or death is agreed on and executed by both cell types. Thus, the toxic and protective properties of microglia may have evolved to increase the overall fitness of the entire organism and promote survival. However, one might speculate that this bidirectional neuronal–glial communication system is compromised in disease states so that the toxic properties of overactive microglia overwhelm neighbouring neurons and lead to their unnecessary destruction. Thus, experimental manipulations that interfere with neuronal–glial intercellular communication can be either neurotoxic or neuroprotective in various disease models, as described further below.
One piece of evidence favouring bidirectional neuronal–microglial communication is the presence of numerous types of neurotransmitter receptors on microglia. As neurons generally release neurotransmitters during synaptic activity, this finding suggests that interneuronal communication can also have indirect effects on the neighbouring microglia that surround the active synapse. Furthermore, if Agnati and Fuxe's classic concept of long-distance communication through ‘volume transmission’5 can be applied to microglia, one might speculate that even those microglia far away from the active synapse may be the recipients of neuroactive signals that have diffused over a considerable distance. This raises the possibility that microglia respond to neurotransmitters from near and far, collect and synthesise this information and communicate back to neurons in a large-scale feedback loop.
Microglia are well known to express receptors for glutamate as well as many other neurotransmitters.6–8 In response to neurotransmitter binding, microglia may increase or decrease their release of neuroactive molecules as part of a positive or negative feedback loop, respectively. The molecules released by microglia in response to neurotransmitter stimulation include free radicals such as superoxide and nitric oxide as well as chemokines and cytokines such as interleukins (ILs) and tumour necrosis factor-α (TNFα), all of which are well known to exert profound effects on neurons. For example, microglial neurotransmitter receptors are known to trigger superoxide production by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and this can be either neuroprotective or neurotoxic depending on the neurotransmitter that originally stimulated the microglial receptor.9 The bidirectional flow of communication between neurons and microglia is especially well reflected in the tight coupling of the activation of neuronal ion channels to microglial activation.10 In general, the ‘on’ signal is hypothesised to trigger receptor-mediated microglial activation, whereas the ‘off’ signal serves to contain microglia and keeps them in the default, non-activated state.10 Neurotransmitter binding could force microglia down either path depending on the needs of the organism.
Below we summarise some of the evidence in support of neurotransmitter effects on microglia, with special emphasis on glutamate, γ-aminobutyric acid (GABA), norepinephrine, cannabinoid and acetylcholine receptors. It is evident from the summary below that there are numerous examples of microglia engaging in either protective or toxic behaviours.
Glutamatergic microglial receptors
Glutamate receptors are classified by their binding to well-studied ligands and can be divided into ionotropic or metabotropic receptors. Ionotropic glutamate receptors are cation-specific ion channels, whereas metabotropic glutamate receptors are a family of G protein-coupled, seven transmembrane domain receptors that impact second messenger systems as well as ion channels. Ionotropic glutamate receptors are classified into α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), kainate or N-methyl-D-aspartate (NMDA) subtypes according to their pharmacological binding properties. Metabotropic glutamate receptors are divided into three groups (groups I, II and III) and eight subtypes (mGlu1—8) based on their pharmacological characteristics, amino acid sequence similarities and downstream signal transduction cascades. Group I includes mGlu1 and mGlu5, group II includes mGlu2 and mGlu3 and group III includes mGlu4, mGlu6, mGlu7 and mGlu8. The microglial response to glutamate is complex and mediated by ionotropic and metabotropic receptors.
The ionotropic glutamate receptors on microglia include AMPA-type GluR1–GluR4 receptors as well as kainate receptors.6 The AMPA receptors on microglia inhibit TNFα release, whereas glutamate and kainate increase TNFα release.11 However, some studies have noted that activation of AMPA receptors on microglia can also stimulate TNFα release.6 The impact of TNFα release from microglia is discussed further below. There is also evidence supporting the presence of NMDA receptors on microglia. For example, microglia are transiently activated following injections of NMDA into the cortex of neonatal rats.12 Furthermore, NMDA receptor subunits have been identified on microglia, and these receptors may enhance the release of TNFα, IL-1 and nitric oxide.13
Microglia express members of all three groups of metabotropic glutamate receptors.14–16 For example, cultured microglia express group I mGlu5a receptor protein, and activation of this receptor reduces microglial TNFα production.14 Microglia also express group II mGlu2 and mGlu3 receptor messenger RNA (mRNA) and protein, and these receptors are both negatively coupled to adenylate cyclase.15 The group II receptors are known to activate microglia, leading to enhanced staining for the ED1 microglial activation marker.16
Group II microglial glutamate receptors are indirectly activated by β-amyloid and chromogranin A, both of which accumulate in senile plaques in Alzheimer's disease. β-Amyloid and chromogranin A elicit the release of microglial glutamate, which, in turn, may bind the group II metabotropic glutamate receptors in microglia in an autologous feedback loop and enhance their toxicity.15 Thus, agonists of group II receptors are thought to induce a neurotoxic phenotype in microglia, whereas antagonists of these receptors have been shown to blunt chromogranin A-induced microglial reactivity and neurotoxicity.15
Microglia also express group III receptors mGlu4, mGlu6 and mGlu8, but not mGlu7.16 In contrast with group II receptors, the activation of group III mGlu receptors induces mild activation of microglia—as evidenced by enhanced ED1 staining—but this activation is not neurotoxic.16 Activation of group III mGlu receptors prevents microglial release of glutamate,8 reduces microglial reactivity in response to lipopolysaccharide (LPS) and β-amyloid and prevents microglial toxicity towards neurons.16 These latter findings suggest that activation of group III mGlu receptors may serve as a potentially therapeutic means to blunt neuroinflammation. Taken together, the studies on metabotropic glutamate receptors suggest that the impact of mGlu receptor activation on microglial reactivity and neurotoxicity depends on the receptor subtype; group II receptor activation may be toxic, but group III receptor activation may be protective (figure 1).
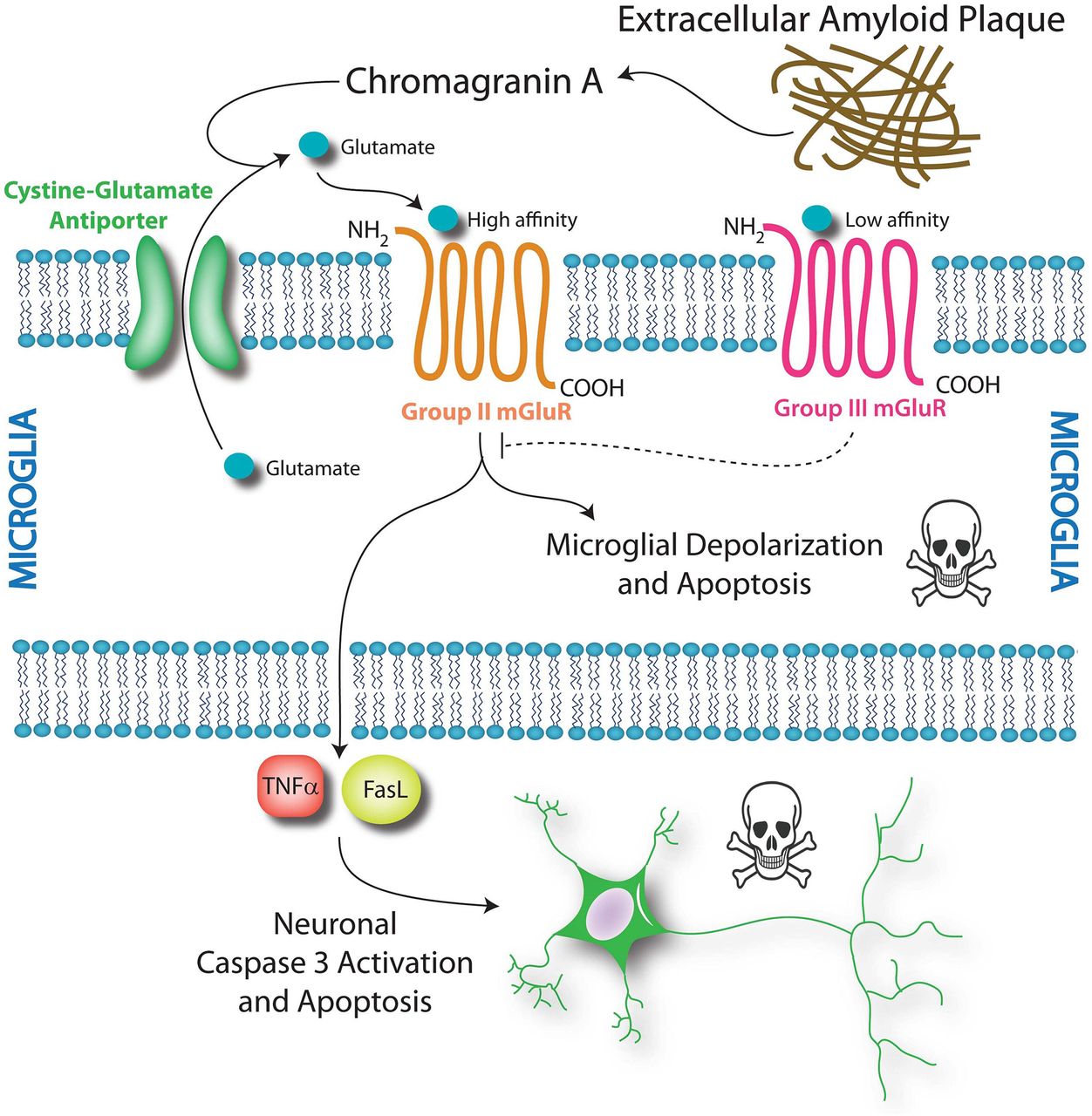
Metabotropic glutamate receptors impact microglial and neuronal survival. Chromogranin A from senile plaques is hypothesised to enhance the release of glutamate from microglial stores via the cystine–glutamate antiporter. This glutamate may bind to high-affinity group II mGluRs and initiate microglial cell death via depolarisation and apoptosis. A second effect of group II mGluR activation is microglial toxicity towards neurons. Activation of the group II mGluR receptor induces tumour necrosis factor-α (TNFα) release from microglia. This TNFα is neurotoxic in the presence of Fas ligand (FasL), which is also derived from microglia. These two soluble molecules stimulate caspase-mediated neuronal apoptosis via the TNF receptor TNFR1. In contrast, activation of the low-affinity group III mGlu receptors does not induce microglial apoptosis. Instead, group III activation reduces microglial reactivity in response to chromogranin A and attenuates microglial toxicity towards neurons. Thus, modulation of microglial metabotropic glutamate receptors may be a pharmacological means to control microglial activation and impact neuronal survival.
Despite the above-mentioned observations on group II metabotropic receptors, activation of group II receptors in microglia is not inevitably toxic. For example, intrastriatal injections of an agonist for group II receptors, DCG-IV ((2S,2′R,3′R)-2-(2′3’-dicarboxycyclopropyl)glycine), markedly activate microglia but nevertheless protect dopaminergic terminals against the parkinsonian toxicant 1-methyl-4-phenylpyridinium (MPP+).17 Striatal cells in this model also upregulate brain-derived neurotrophic factor (BDNF). Although DCG-IV protects dopamine neurons, it is actually toxic to cells in the striatum.18 These findings reveal that microglial activation may be associated with diverse effects on various cell types. In sum, the impact of glutamatergic receptor activation on microglia appears to vary depending on receptor subtype, cellular phenotype as well as the type of stimulus (such as MPP+ vs chromogranin A).
As with ionotropic receptors, other effects of metabotropic glutamate receptor activation in microglia include changes in TNFα release.6 ,11 Although activation of group I receptors reduces TNFα production in cultured microglia,14 stimulation of mGlu2 triggers TNFα-induced neurotoxicity associated with the Fas death receptor ligand.19 Fas ligand released in this manner activates caspase 3 in neurons via p55 and thereby elicits neuronal death (figure 1). Under these conditions, TNFα is only neurotoxic in the presence of microglia or microglia-conditioned medium because Fas ligand must also be present.19 Thus, glutamate receptor activation in microglia may kill neighbouring neurons in a TNFα/Fas ligand-dependent and caspase-dependent manner. It should be noted, however, that TNFα and IL-6 may also be neuroprotective, particularly under ischaemic conditions.20 ,21 Therefore, the impact of glutamate receptor activation on microglia might have more subtle and varied effects than anticipated.
GABAergic microglial receptors
GABA is the major inhibitory transmitter in the central nervous system and binds to one of the two types of receptors—GABAA and GABAB. GABAA receptors are ionotropic, whereas GABAB receptors are metabotropic. Both groups of receptors are divided into subtypes. Microglia express all three subtypes of GABAB receptors in culture and in vivo, supporting the hypothesis that microglia are cellular targets for the GABA neurotransmitter.22 There is some evidence to suggest that these receptors modulate the release of proinflammatory microglial cytokines such as ILs in response to GABAergic tone. For example, GABAB receptor agonists trigger the induction of an outwardly rectifying K+ conductance in microglia and attenuate the LPS-induced release of microglial cytokines such as IL-6 and IL-12p40.22 In contrast, the GABAB receptor did not affect TNFα or nitric oxide secretion in these studies.
It is noteworthy that microglia increase their expression of GABAB receptors in response to injuries.22 Given the GABAB receptor-induced attenuation of microglial IL secretion, this compensatory increase in receptors during times of stress may reflect the generally protective role of the inhibitory GABA neurotransmitter. This view is consistent with reports that GABA promotes a neuroprotective phenotype in microglia.9
Microglia also express GABAA receptors. GABAA receptors trigger microglial superoxide production and promote a neuroprotective microglial phenotype.9 The GABAA receptor agonist muscimol inhibits the LPS-induced release of TNFα and IL-6 from microglia.23 Microglia may also indirectly respond to muscimol, as this GABAA agonist stimulates neuronal and macroglial GABAA receptors and increases extracellular K+ concentrations. This increase in K+, in turn, causes microglia to release chemokines such as macrophage inflammatory protein-1α (MIP1α).24 Thus, microglia can respond directly and indirectly to GABAergic tone.
Norepinephrine microglial receptors
Catecholamines such as norepinephrine and epinephrine bind to G protein-coupled adrenergic receptors, usually eliciting a sympathomimetic response. There are two main groups of adrenergic receptors, α and β, and each group contains multiple subtypes with homologous sequences. The α group contains α1 and α2 subtypes, which increase inositol triphosphate and diacylglycerol or decrease cyclic AMP, respectively. The β group contains β1, β2 and β3 subtypes and is generally linked to activation of adenylate cyclase and subsequent increases in intracellular cyclic AMP. Many research groups have established the presence of adrenergic receptors on microglia.25 Norepinephrine and β1-adrenergic agonists generally increase cyclic AMP levels in microglia.26 ,27 Therefore, it is not surprising that forskolin, a cyclic AMP-elevating agent, can mimic some of the effects of β-adrenergic agonists on microglia.27
Microglial cells express mRNAs encoding α1A, α2A, β1 and β2 receptors.26 Agonism at these receptors appears to dampen microglial reactivity. For example, the toxicity of microglia can be downregulated by β-adrenergic agonists.25 Furthermore, norepinephrine and β1-adrenergic agonists suppress the production of proinflammatory cytokines such as IL-6 and TNFα.26 ,28 In line with the dampening of microglial activation, catecholamines also inhibit nitric oxide production from microglia, perhaps by causing a decrease in inducible nitric oxide synthase.28 ,29 These findings reveal that free radical production from microglia is attenuated by the action of catecholamines on adrenergic receptors. Owing to their attenuation of free radical and cytokine release, adrenergic agonists present a means to mitigate the cytotoxicity of microglia. It is also noteworthy that depletion of adrenergic tone negatively affects the microglial phagocytic clearance of β-amyloid.30 Thus, catecholaminergic tone in the brain may modulate inflammation in a protective way.
Cannabinoid microglial receptors
Cannabinoid receptors are G protein-coupled receptors that bind to tetrahydrocannabinol (THC), the major psychoactive ingredient in marijuana. These receptors have evolved to respond to endocannabinoids, endogenous ligands that include anandamide. Cannabinoid receptors are divided into two groups, CB1 and CB2. It is the CB2 receptor subtype that is primarily expressed in microglia.31 Endocannabinoids can regulate microglial reactivity by binding to CB2 and inhibiting adenylate cyclase.32 This microglial receptor is overexpressed in neuroinflammatory disorders, as discussed below.31 CB2 receptor activation stimulates microglial migration but inhibits the release of cytokines such as IL-6 and TNFα.32 Thus, it is not surprising that CB2 agonists have been shown to be protective in several experimental models of diseases. For example, the CB2 agonist JWH-015 improves motor function under the demyelinating conditions induced by Theiler's murine encephalomyelitis virus.33 Another CB2 agonist, JWH-133, significantly decreases the immunostaining intensity of Iba1+ activated microglia and blunts the expression of proinflammatory factors such as IL-6, TNFα and inducible nitric oxide synthase (iNOS) in a mouse model of stroke. These effects are reversed by inhibition of CB2R or absent in CB2R knockout mice.34 Pretreatment with AM1241, another CB2 agonist, has also been reported to attenuate LPS plus interferon γ (IFNγ)-induced microglial activation by shifting microglial polarisation from the toxic M1 state to the protective M2 state.35 In models of Alzheimer's disease, the endocannabinoid system may be similarly protective. For example, the CB1/CB2 agonist WIN55 inhibits β-amyloid-mediated activation of microglia in vitro and β-amyloid-mediated toxicity in vivo.36 Furthermore, overexpression of CB2R reduces the immunostaining intensity of Iba1+ activated microglia and results in neuroprotection against the 6-hydroxydopamine model of Parkinson's disease.37
Given the above-mentioned findings that CB2 receptors may be protective against multiple disease states, it is not surprising that CB2 knockout animals are, conversely, more vulnerable in several models of human diseases. For example, CB2 gene ablation increases 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) toxicity in models of Parkinson's disease.38 CB2 knockout animals also show increased microglial activation and greater axonal loss in the inflamed spinal cord.39 The endocannabinoid system is thus hypothesised to counteract the progression of disorders such as Huntington's disease, multiple sclerosis, amyotrophic lateral sclerosis and stroke. Perhaps because of its intrinsically protective properties, this system appears to be upregulated under conditions of injury, perhaps as a compensatory response to preserve homeostasis.32 For example, upregulation of CB2R has been reported in rat models of Parkinson's disease.40 In humans suffering from multiple sclerosis, CB2 expression is increased in the microglia associated with white matter.41 The CB2 receptor is also increased postmortem tissue in amyotrophic lateral sclerosis.41 In short, the endocannabinoid system may be upregulated in disease states in order to help the brain adapt to pathological stress. This is consistent with reports that activation of CB2R induces an anti-inflammatory phenotype in microglia.37
Acetylcholine microglial receptors
Acetylcholine receptors are classified as nicotinic or muscarinic. Nicotinic receptors are ionotropic cation channels permeable to Na+ and K+ and subdivided into muscle-type and neuronal-type (α2–α10 and β2–β4) receptors. Muscarinic receptors are metabotropic G protein-coupled receptors, some of which deactivate adenylate cyclase and activate K+ channels (M2, M4), while others upregulate phospholipase C, inositol triphosphate and intracellular calcium (M1, M3, M5). Nicotinic α3, α5, α6, α7 and β4 receptors are expressed in microglia.42 ,43 Muscarinic receptors are also expressed in cultured microglia, but far less is known about their function.44
The nicotinic α7 receptors on microglia increase intracellular calcium through the inositol triphosphate pathway.45 Nicotine increases or decreases microglial TNFα release in response to purinergic receptor activation or LPS stimulation, respectively.42 ,45 The inhibition of TNFα release from microglia by acetylcholine or nicotine is mediated by a reduction in ERK1/2 and p38 MAP kinase signalling.42 In general, nicotinic receptors dampen microglial activation. For example, nicotine reduces free radical production from microglia in response to β-amyloid.46 Nicotinic receptor activation in microglia also inhibits their activation in response to IFNγ.47 Furthermore, activation of nicotinic α7 receptors induces an increase in antioxidant genes and a decrease in the phosphorylation of nuclear factor kappa B (NF-κB) and p65 in microglia, and promotes microglial M2 polarisation.48 All of these findings support the view that acetylcholine dampens the immune response of the brain.
Other receptors of neurotransmitters and conclusion
Other types of microglial receptors not covered in the present review include receptors for bradykinin,49 dopamine,28 purine,49 adenosine49 and opioids,49 among other transmitters. Through these multiple types of neurotransmitter receptors, neurons can actively control microglial function, often, but not always, dampening microglial activation. Conversely, microglia actively control neuronal function in a feedback loop. For example, it has long been known that microglia modulate synaptic plasticity by the secretion of inflammatory mediators, triggering changes in learning and memory and affecting adaptive behaviour.50 Given the plasticity of the brain and its inherent ability to respond to challenges, it is not surprising that the flow of communication between these two fundamental cell types is in both directions. As argued earlier, this loop may be part of a phylogenetically ancient design that allows microglia to respond dynamically to the synaptic release of neurotransmitters and control neuronal state to promote survival and organismal fitness.
In summary, microglia are typically quiescent under physiological, healthy conditions but can be rapidly activated under pathological conditions, exerting either toxic or neuroprotective effects in accordance with their polarisation phenotypes. The quiescent state of microglia is maintained, at least partly, by neurotransmitters released from neurons.3 For example, a decrease in morphological change and process motility has been observed in retinal microglia after activation of the GABAA receptor.51 On the other hand, pathological neuronal activity may contribute to the inflammatory milieu of the brain because of the presence of neurotransmitter receptors on microglia. For example, during pathological processes, neurons may release sufficient glutamate to initiate an excitotoxic cascade and activate neighbouring microglia.52 Besides exacerbating microglial inflammation and neurotoxicity, activation of microglial neurotransmitter receptors also may induce anti-inflammatory effects by reducing TNFα42 and IL-6,22 decreasing superoxide53 and nitric oxide14 production, and thereby eliciting neuroprotective effects. The functional roles of microglial neurotransmitter receptors in the maintenance of microglial quiescence or phenotypic switches are still poorly understood. Also, we have very little information about the interactions between different microglial receptors and their combined downstream effects. Different microglial neurotransmitter receptors might be simultaneously or sequentially activated (figure 2). Although these receptors have been individually studied, the interplay between them remains unknown. In addition, the lack of a specific antibody recognising specifically either microglia or macrophages precluded the possibility of distinguishing between the local microglia and circulating macrophages that were recruited to the injured brain. As a result, many studies regarding the function of microglial neurotransmitter receptors in the injured brain may also include the infiltrated macrophages. Further research to elucidate the role of microglial receptors and test their therapeutic potential in neurological disorders is urgently needed.54

{kind=link}
{kind=link}
Different effects of various microglial neurotransmitter receptors. AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; cAMP, cyclic adenosine monophosphate; ERK, extracellular signal-regulated kinase; FasL, Fas ligand; GABA, γ-aminobutyric acid; iNOS, inducible nitric oxide synthase; IFNγ, interferon γ; IL-1, interleukin 1; IL-6, interleukin 6; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; NMDA, N-methyl-D-aspartate; NO, nitric oxide; TNFα, tumour necrosis factor-α.
References
Footnotes
Contributors HL, RKL and XH wrote the manuscript.
Funding This work was supported by the NIH/National Institute of Neurological Disorders and Stroke (NINDS) grants NS092618 (to XH), a grant (13SDG14570025) from the American Heart Association (to XH).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.