Article Text

Abstract
Rationale Haematoma growth is common early after intracerebral haemorrhage (ICH), and is a key determinant of outcome. Tranexamic acid, a widely available antifibrinolytic agent with an excellent safety profile, may reduce haematoma growth.
Methods and design Stopping intracerebral haemorrhage with tranexamic acid for hyperacute onset presentation including mobile stroke units (STOP-MSU) is a phase II double-blind, randomised, placebo-controlled, multicentre, international investigator-led clinical trial, conducted within the estimand statistical framework.
Hypothesis In patients with spontaneous ICH, treatment with tranexamic acid within 2 hours of onset will reduce haematoma expansion compared with placebo.
Sample size estimates A sample size of 180 patients (90 in each arm) would be required to detect an absolute difference in the primary outcome of 20% (placebo 39% vs treatment 19%) under a two-tailed significance level of 0.05. An adaptive sample size re-estimation based on the outcomes of 144 patients will allow a possible increase to a prespecified maximum of 326 patients.
Intervention Participants will receive 1 g intravenous tranexamic acid over 10 min, followed by 1 g intravenous tranexamic acid over 8 hours; or matching placebo.
Primary efficacy measure The primary efficacy measure is the proportion of patients with haematoma growth by 24±6 hours, defined as either ≥33% relative increase or ≥6 mL absolute increase in haematoma volume between baseline and follow-up CT scan.
Discussion We describe the rationale and protocol of STOP-MSU, a phase II trial of tranexamic acid in patients with ICH within 2 hours from onset, based in participating mobile stroke units and emergency departments.
- stroke
- hemorrhage
- CT
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Intracerebral haemorrhage (ICH) causes approximately 3 million deaths per year worldwide. This is a similar number of deaths to ischaemic stroke despite ischaemic stroke being twice as common.1 While there are a number of highly effective treatment options for ischaemic stroke, therapies for ICH remain extremely limited. Organised stroke unit care reduces mortality and dependency in ICH.2 Additionally, early blood pressure lowering to a target of <140 mm Hg may improve functional outcomes,3 but more aggressive blood pressure lowering has not been shown to be beneficial.4 The role of surgical intervention remains limited and not well established.5
Haematoma growth is one of the strongest predictors of morbidity and mortality after ICH and is a key therapeutic target (figure 1).6 Acute medical interventions that attenuate haematoma expansion have been proposed as a potential therapeutic strategy. One of these, recombinant activated factor VII (rFVIIa) has consistently reduced haematoma growth in phase II and III trials, but has been associated with an increase in thromboembolic complications.7 8 The high cost of this agent also limits access in many healthcare settings. Tranexamic acid, an antifibrinolytic agent, is inexpensive, widely available and has an excellent safety profile. It has been shown to improve survival and outcome in haemorrhage due to acute trauma,9 and specifically in the setting of traumatic brain injury when administered within 3 hours.10 It has also been shown to reduce rebleeding following subarachnoid haemorrhage,11 although this has not translated to improved outcomes in this condition.11 12
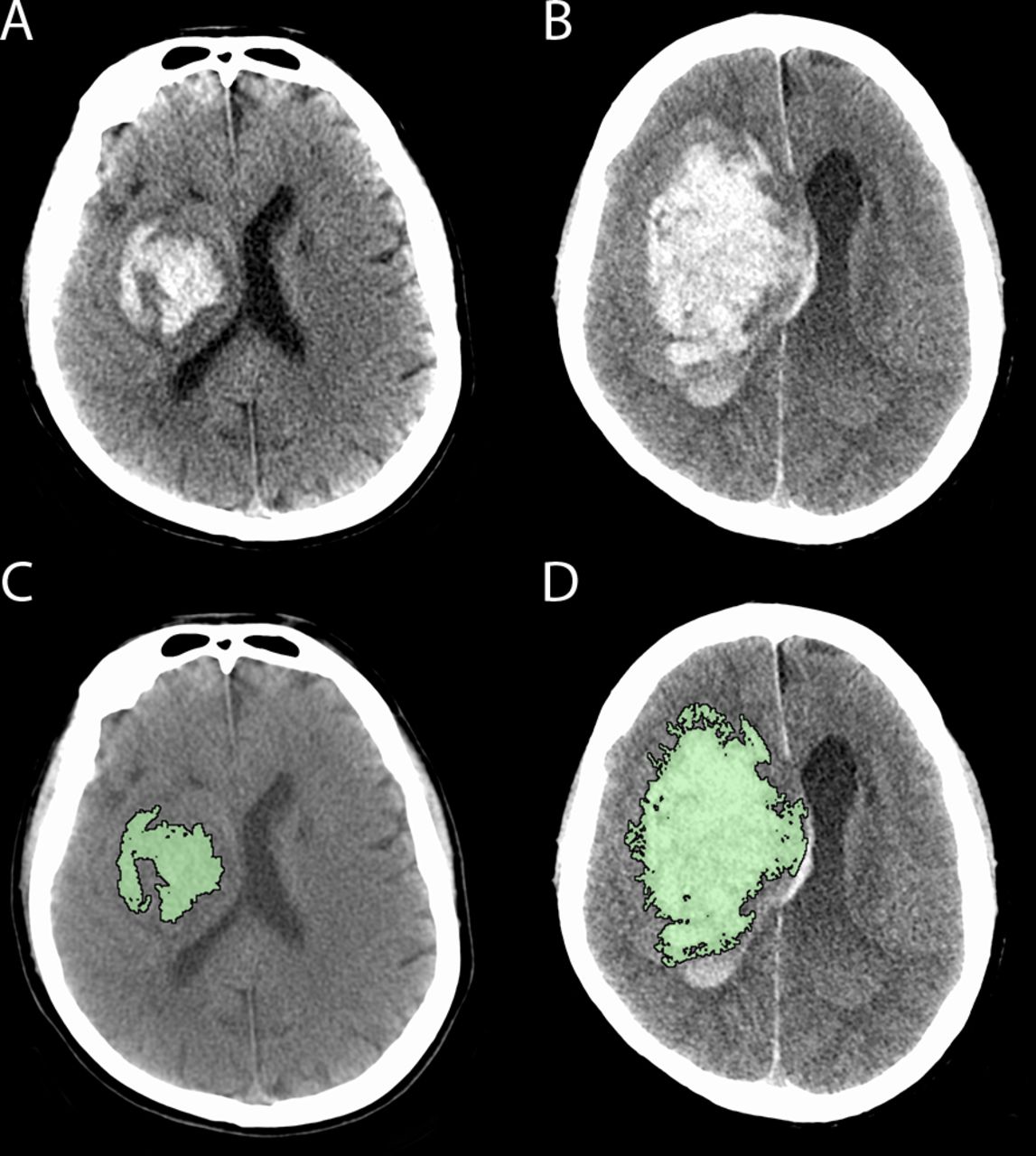
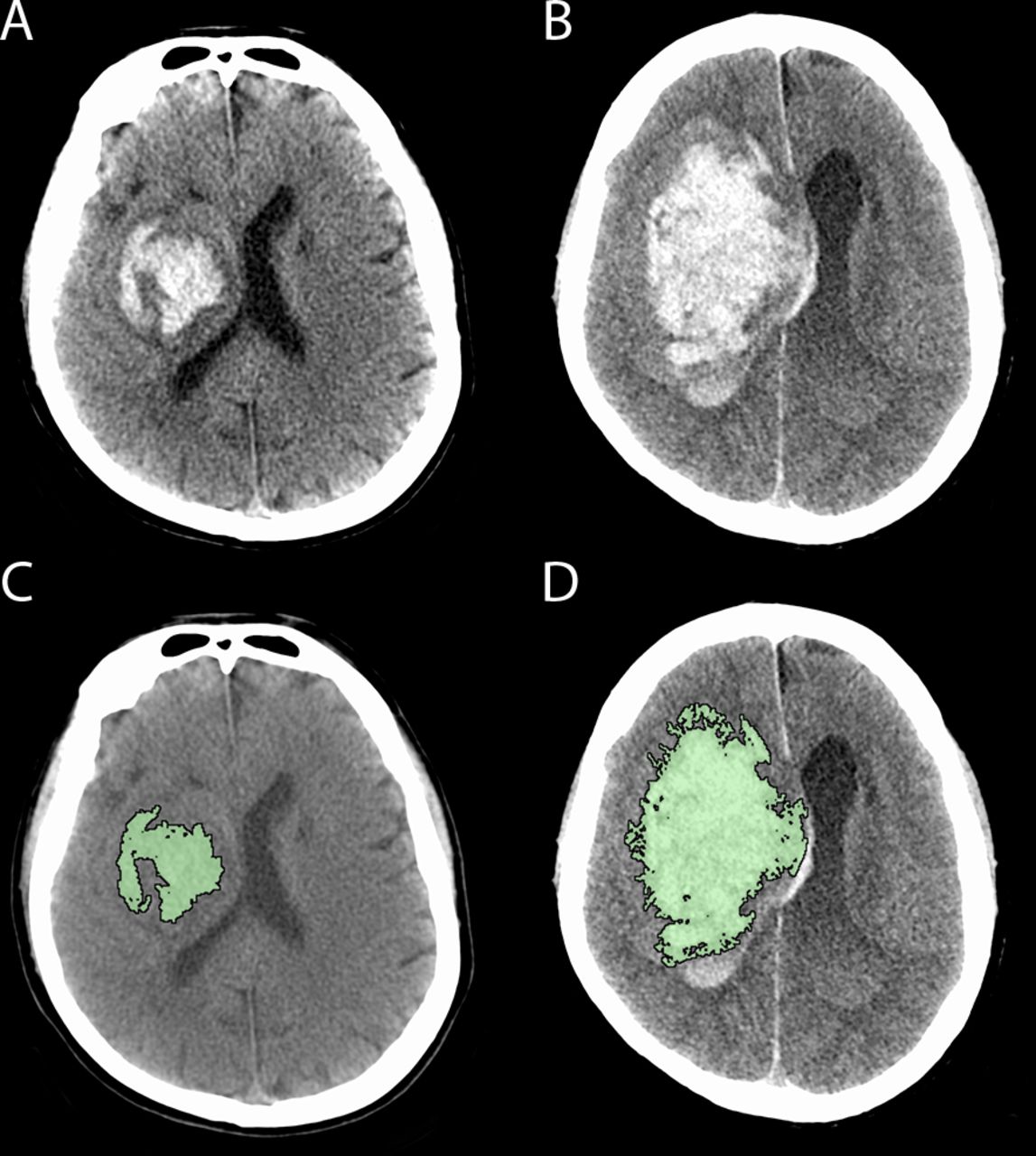
Haematoma growth occurring between baseline (A, C) CT and follow-up CT (B, D) in a patient with a right basal ganglia haemorrhage. Panel C and D reflect the region of interest planimetric analysis used to measure haematoma volume (intraventricular segment measured separately).
Tranexamic acid modestly reduced ICH expansion in the tranexamic acid for hyperacute primary intracerebral haemorrhage (TICH-2) trial, which included participants up to 8 hours from symptom onset, and was also associated with a lower mortality at 7 days.13 However, the recently published tranexamic acid for acute ICH growth predicted by spot sign (TRAIGE) trial did not show a significant reduction in haematoma growth with tranexamic acid in patients treated up to 8 hours from onset who had imaging features of ongoing bleeding, such as the CT angiography ‘spot-sign’. The median onset to treatment time in this trial was 4 hours and 50 min.14 In the phase II spot sign and tranexamic acid on preventing ICH growth—Australasia (STOP-AUST) trial, there was a trend towards reduced haematoma growth in patients treated within 4.5 hours of onset who had the spot sign, with the strongest trends seen in those in the prespecified subgroup treated within 3 hours of onset, as well as a post-hoc subgroup treated within 2 hours of onset.15
The presence or absence of a spot sign imperfectly predicts ICH expansion16 and ultraearly ICH detected on imaging is itself a predictor of subsequent expansion.17 The benefits of ultraearly intervention for ischaemic stroke has led to patients who had a stroke receiving ever-earlier imaging postsymptom onset, increasingly in a mobile stroke unit (MSU) setting. Therefore, selecting patients with non-contrast CT in an ultraearly time frame may both provide the optimal time window for intervention and maximise generalisability. MSUs facilitate this unique opportunity for ultraearly assessment and management of ICH.18
In this paper, we outline the protocol of the stopping intracerebral haemorrhage with tranexamic acid for hyperacute onset presentation including mobile stroke units (STOP-MSU) trial, a prospective, phase II, randomised, double-blind, placebo-controlled, investigator-driven, multicentre, international clinical trial which will be performed in patients with ICH in both prehospital and in-hospital settings. The study objective is to determine the efficacy and safety of intravenous tranexamic acid in patients with ICH within 2 hours of onset.
Methods
The main hypothesis of STOP-MSU is that in patients with primary ICH, treatment with tranexamic acid within 2 hours of onset will reduce haematoma expansion compared with placebo. Key trial activities are summarised in figure 2.

{kind=link}
{kind=link}
Key trial timepoints and activities. ICH, intracerebral haemorrhage; mRS, Modified Rankin Scale; NIHSS, National Institutes of Health Stroke Scale; TXA, tranexamic acid.
The study has been prospectively registered on clinicaltrials.gov (NCT03385928) and will be conducted within the estimand framework for the design and analysis of clinical trials. The objective of the estimand framework as presented in addendum (R1) to the International Council on Harmonization E9 guidance,19 is to align the clinical trial objectives with the study design, endpoints, and analysis, in order to improve study planning and the interpretation of the study results. The trial design and estimands include prespecified strategies for the conduct of the trial during the COVID-19 pandemic.20–23
Patient population
The trial will include patients with acute primary ICH confirmed on non-contrast CT, who are aged ≥18 years, are eligible for recruitment, and able to be treated with the investigational product within 2 hours of stroke onset. Patients will be recruited from both MSUs and participating emergency departments. Inclusion and exclusion criteria are outlined in box 1.
Key inclusion and exclusion criteria
Inclusion criteria
Patients presenting with an acute ICH confirmed on non-contrast CT
Age≥18 years
Treatment can commence within 2 hours of symptom onset (or in patients with unknown time of symptom onset, the time the patient was last known to be well)
Consent can be obtained from the participant or a person responsible. Oral consent is allowed in some jurisdictions. When emergency treatment procedures have been followed (where permittable) the participant or person responsible will be asked for consent to continue in the study.
Exclusion criteria
Glasgow Coma Scale total score of <8
Brainstem ICH
ICH volume >70 mL as measured by the ABC/2 method
ICH known or suspected by study investigator to be secondary to trauma, aneurysm, vascular malformation, haemorrhagic transformation of ischaemic stroke, cerebral venous thrombosis, thrombolytic therapy, tumour, or infection
Any history or current evidence suggestive of venous or arterial thrombotic events within the previous 90 days, including clinical, ECG, laboratory, or imaging findings. Clinically silent chance findings of old ischaemia are not considered an exclusion.
Hereditary or acquired haemorrhagic diathesis or coagulation factor deficiency.
Use of heparin, low-molecular weight heparin, GPIIb/IIIa antagonist, or oral anticoagulation (eg, warfarin/vitamin K antagonists, factor Xa inhibitor, thrombin inhibitor) within the previous 72 hours. Warfarin/vitamin K antagonists must also have an International Normalised Ratio of <1.3 prior to randomisation.
Pregnancy (women of childbearing potential must be tested)
Planned surgery for ICH within 24 hours
Concurrent or planned treatment with haemostatic agents (eg, prothrombin complex concentrate, vitamin K, fresh frozen plasma, or platelet transfusion)
Participation in any investigational study in the last 30 days
Known terminal illness or planned withdrawal of care or comfort care measures.
Any condition that, in the judgement of the investigator could impose hazards to the patient if study therapy is initiated or affect the participation of the patient in the study.
At the time of writing, the study is being conducted in one MSU (the Melbourne MSU), as well as 21 other hospital sites in Australia, New Zealand, Taiwan and Finland, including two sites in rural/regional areas. The Melbourne MSU operates in central metropolitan Melbourne with an approximate catchment population of 2 million people. The operational details have been described previously.24 Briefly, the service is staffed by a neurologist or senior stroke fellow, stroke advanced practice nurse, CT radiographer, advanced life support paramedic and mobile intensive care paramedic. The MSU incorporates a self-mobile CereTom CT scanner (Samsung Neurologica Corp, Massachusetts, USA). Participants recruited on the MSU undergo enrolment, randomisation and treatment administration on the MSU and are subsequently transported to a participating hospital site, where remaining study activities are performed.
Consent
Depending on local legal and regulatory provisions at each site, consent can be obtained in either written or oral form from the participant or person responsible. In some jurisdictions, recruitment into the trial under emergency treatment provisions is approved. In these cases, the participant or person responsible will be subsequently approached to provide consent to continue in the study. Where there has been rapid clinical deterioration leading to death or institution of palliative care, the Human Research Ethics Committee has granted a waiver of the requirement for consent to continue as this may cause significant distress to the person responsible and may risk introducing systematic bias by excluding participants with poor clinical outcomes.
Randomisation
This is a double-blind trial. The patient and all those involved in the clinical and imaging assessment of outcomes will be blinded to treatment allocation. Patients will be randomised using a blinded prerandomised assignment procedure in order to minimise potential communication-related delays (either blinded treatment packs or envelopes depending on the site). Randomisation will be in a 1:1 ratio to receive either tranexamic acid or placebo stratified by treating centre and using randomly permuted blocks. The treatment allocation table has been prepared by the study statistical team and is not available to the investigators.
Treatment or intervention
At most centres, the investigational product will be distributed in prerandomised externally indistinguishable sealed treatment packs that contain either tranexamic acid or 0.9% saline in non-identical vessels. After enrolment, the lowest numbered sealed treatment pack will be handed to an external unblinded person (a person not involved in patient management or evaluation, such as a paramedic or emergency department nurse, based on local centre practices) who will give 1 g over 10 min as per local protocol; followed by 1 g over 8 hours as per local protocol. For sites where packaged kits are not acceptable, randomisation envelopes will be provided. After enrolment, the lowest numbered randomisation envelope will be opened by an external unblinded person (a person not involved in patient management or evaluation, such as a paramedic or emergency department nurse, based on local centre practices) who will open the randomisation envelope out of sight of the patient and study personnel and obtain the appropriate ampoules based on the randomisation envelope contents. They will give 1 g over 10 min as per local protocol; followed by 1 g over 8 hours as per local protocol. The volume of 0.9% NaCl diluent will be based on local protocol.
Investigations including electrocardiography and standard baseline laboratory investigations (haematology, clinical chemistry, coagulation studies) will be performed and collected as part of routine clinical care. No additional laboratory investigations will be performed on the MSU as part of the trial.
Study estimands
Primary estimand
Population: all included participants.
Individual-level outcome measure: presence or absence of intracerebral haematoma expansion by 24±6 hours as defined by either ≥33% or≥6 mL increase from baseline on CT scan. Haematoma volume will be measured on non-contrast CT using a validated semi-automated planimetric method (figure 1).25
Population level measure: proportion of participants with outcome present in each group.
Strategy for intercurrent events: composite strategy
Any patient related intercurrent event that prevents collection of the primary outcome measure will result in the outcome being imputed as growth present. This typically relates to participant clinical deterioration and a decision being made for palliative care prior to collection of the primary outcome measure. In cases where an intercurrent event (patient or system-related) leads to early collection of the primary outcome (ie, <18 hours), growth will be imputed if it is observed as occurring despite the early measurement. In cases where an intercurrent event (patient or system-related) leads to delayed collection of the primary outcome (>30 hours but <36 hours), absence of growth will be imputed if growth is not observed on the delayed assessment.
Any system-related intercurrent events which prevent collection of the primary outcome measure according to the rules specified above will be examined for missingness at random and standard strategies for dealing with missing data26 will be employed as specified in the statistical analysis plan (SAP).
Secondary estimand—growth
Population: all included participants without patient or system related intercurrent events preventing measurement of haematoma volume at 24±6 hours.
Individual level outcome measure
Supplementary estimand 1: haematoma growth—alternate definition: ≥33% or ≥6 mL increase in intracerebral haematoma volume or any increase in intraventricular volume by 24±6 hours.
Supplementary estimand 2: absolute intracerebral haematoma growth
Supplementary estimand 3: relative intracerebral haematoma growth
Supplementary estimand 4: absolute intraventricular haematoma growth
Supplementary estimand 5: absolute intracerebral plus intraventricular haematoma growth.
Population level measure:
Supplementary estimand 1: proportion of participants with outcome present in each group
Supplementary estimands 2–5: difference in medians between groups for each supplementary estimand, adjusted for baseline haematoma volume.
Strategy for intercurrent events:
Supplementary estimand 1: composite strategy as for primary estimand
Supplementary estimands 2–5: principal stratum strategy.
Any patient related intercurrent event that prevents collection of the primary outcome measure will result in the patient being excluded from this analysis. This typically relates to patient clinical deterioration and a decision being made for palliative care prior to collection of the follow-up imaging. If any intercurrent event (patient or system related) leads to early or delayed collection of the imaging follow-up outcome (ie, <18 hours or >30 but <36 hours), these participants will still be included in the secondary estimand analysis, but a sensitivity analysis will be conducted on the population where imaging was conducted strictly within 18 to 30 hours (sensitivity estimands within each supplementary estimand)
Secondary estimand—functional outcome
Population: all included participants.
Individual level outcome measure
Supplementary estimand 1: Modified Rankin Scale (mRS)<3 or equal to pre-stroke baseline at 90 days (absent or present)
Supplementary estimand 2: mRS <4 or equal to pre-stroke baseline at 90 days (absent or present)
Supplementary estimand 3: mRS at 90 days
Supplementary estimand 4: utility-weighted mRS at 90 days.
Population level measure
Supplementary estimand 1: proportion of participants with outcome present in both groups
Supplementary estimand 2: proportion of participants with outcome present in both groups
Supplementary estimand 3: ordinal comparison of mRS across all levels (mRS 5–6 combined) between groups
Supplementary estimand 4: mean utility weighted mRS score in both groups.
Strategy for intercurrent events: treatment policy
While COVID-19 presents a unique risk of intercurrent death (mRS=6) or functional impairment due to the potential for increased incidence over the study period, we will include patients with proven COVID-19 infection in the functional outcome estimands. Additionally, we will perform a sensitivity estimand analysis excluding those with confirmed COVID-19 infection.
Secondary estimand—safety—death
Population: all included participants.
Individual-level outcome measure
Death within 3 months (absent or present).
Death within 7 days (absent or present).
Population level measure
Proportion of participants with outcome present in each group.
Strategy for intercurrent events: treatment policy
As with the functional outcome estimand, we will include patients with proven COVID-19 infection in both death outcome estimands. Additionally, we will perform a sensitivity estimand analysis excluding those with confirmed COVID-19 infection.
Secondary estimand—safety—major thromboembolic events
Population: patients without confirmed COVID-19 infection during the study period.
Individual-level outcome measure
Major thromboembolic events (ischaemic stroke, myocardial infarction or pulmonary embolism) within 3 months (absent or present).
Population level measure
Proportion of participants with outcome present in each group.
Strategy for intercurrent events: principal stratum
Given the unique risks of thromboembolic complications posed by COVID-19 infection,27 this estimand will be assessed in those patients in whom confirmed COVID-19 infection did not occur within 3 months of enrolment.
Assessment of haematoma expansion at 1±1 hour will be conducted as exploratory estimands.
All assessments of neurological impairment or functional outcome will be performed by a healthcare professional trained in their administration.
Independent Data Safety Monitoring Board
To compare the safety of tranexamic acid versus placebo, two primary safety variables (mortality and major thromboembolic events) are considered serious enough to warrant inclusion in the interim safety analysis and will be tested independently. This analysis will be undertaken at the completion of the first 40 90-day participant assessments, and subsequently following completion of each 50 90-day participant assessments. If there are concerns about the safety of participants, the independent Independent Data Safety Monitoring Board (DSMB) will make a recommendation to the trial executive committee regarding continuing, stopping or modifying the trial. The Haybittle-Peto procedure for generating early stopping boundaries will be used. A recommendation of early termination due to safety reasons will be considered by the Independent DSMB if the corresponding Haybittle-Peto boundary (p=0.001, Z=3) at a given interim analysis is crossed. No formal interim analyses for efficacy or futility are planned
Sample size estimates
Originally, a sample size of 124 patients was estimated to give 80% power to detect haematoma growth of ≥33% or ≥6 mL in 39% in the placebo arm compared with 14% in the treatment arm, under a two-tailed significance level of 0.05. Adaptive increase in sample size was planned to be performed if the results of interim analysis of the first 75 patients were promising, using the methodology of Mehta and Pocock.28 Following the recruitment of 49 participants, and based on the faster than anticipated recruitment, the trial executive committee made a fully blinded decision to increase the sample size to 180 patients (90 patients per arm) in order to detect an absolute difference in the primary outcome of 20% (placebo 39% vs treatment 19%) under identical power and two-tailed significant level settings. The trial executive committee was satisfied that this smaller effect size still represents a clinically meaningful outcome. Adaptive sample size re-estimation, using the methodology of Mehta and Pocock, will be undertaken based on the outcomes of 144 patients with the possibility to increase the sample size to a prespecified maximum of 326 patients.
Statistical analyses
Statistical analysis will be performed within the estimand framework as described above. The primary efficacy estimand analysis will be performed using a two-sided threshold for statistical significance of 0.05. For the primary estimand analysis, the proportion of patients with ICH growth indicated by either ≥33% or ≥6 mL growth from baseline by 24 hours will be compared between the treatment and control arms of the trial adjusted for baseline haematoma volume using binary logistic regression. Both baseline and 24 hour ICH volumes will be analysed using planimetric techniques at the co-ordinating centre.
Secondary efficacy and safety estimand analyses will also be performed by comparing the treatment and control arms using appropriate regression models: logistic regression for dichotomous outcomes and median regression for continuous outcomes. Analyses involving haematoma growth will be adjusted for baseline haematoma volume as per the primary analysis. Analyses of mRS outcomes (including death) will be adjusted for age and baseline haematoma volume. Analysis for the major thromboembolic event safety estimand will be unadjusted.
Analysis of the categorical shift in functional outcome will be undertaken on the full range of the mRS (mRS 5–6 combined) using proportional odds logistic regression adjusted for age and baseline ICH volume, subject to the validity of shift analysis model assumptions. Should the proportional odds assumption not be satisfied, an assumption-free full scale analysis (mRS 5–6 combined) based on generalised ORs will be used.29
An intention-to-treat analysis will be the main analysis reported for all estimands unless explicitly stated otherwise in the relevant estimand definitions. In addition, a per-protocol analysis will be reported which will be restricted to participants who received at least 50% of the study drug and in whom there were no protocol violations. The details of statistical analysis will be presented in a separate SAP document that will be finalised prior to the study database lock.
Study organisation and funding
The study is run by an executive committee cochaired by SMD and GAD and including HZ and NY as coprincipal investigators and medical coordinators, and LC as trial biostatistician. The study executive committee has access to the complete dataset. In addition, a study steering committee includes all site principal investigators.
Neuroscience Trials Australia (a subsidiary of the Florey Institute of Neuroscience and Mental Health) is the study sponsor. The study is funded by a grant from the Medical Research Future Fund, an initiative of the Australian government Department of Health.
Risk assessment due to COVID-19
In accordance with the guidance documents by the US Food and Drug Administration,20 European Medicines Agency,21 and National Health and Medical Research Council,22 to ensure the safety of study participants, maintain compliance with good clinical practice, and minimise risks to trial integrity due to COVID-19, a risk assessment of trial integrity and conduct was undertaken in July/August 2020. This review considered the potential impacts of the pandemic on the trial within the estimand framework, specifically the potential impacts on the study population, the ability to obtain the outcome measures, the reliability of the outcome measures, and potential COVID-19 related intercurrent events including participants being tested for COVID-19, participants developing confirmed COVID-19 infection, quarantine and travel limitations, site closures, interruption to supply chain of the investigational product or other medications, and delayed or missed visit assessments. The outcomes of this assessment and practical measures to mitigate these risks are outlined in the online supplementary
Summary and discussion
There has been renewed interest in the use of tranexamic acid for emergency management of bleeding in the last decade. In ICH, tranexamic acid has been shown to reduce haematoma expansion with no increase in thromboembolic complications.13 While early evidence supporting rFVIIa is also promising, the costs associated with the therapy are likely to limit its widespread generalisability, particularly in lower income countries, which are known to have a disproportionately high burden of ICH.30 In the Australian context, the cost of a dose of 6 mg of rFVIIa (required for a 75 kg adult) is ~$A8000, compared with a total cost of ~$A10 for 2 g of tranexamic acid.
For any medical therapy aiming to reduce haematoma expansion, inclusion of patients beyond the first few hours after onset, where growth becomes less likely, may limit the opportunity to identify the therapeutic potential of the drug. We therefore chose an ultraearly 2 hour time window to deliver the intervention. This requires highly organised and streamlined acute stroke assessment processes, and we anticipate recruitment on MSUs to be a key element of the trial.
In STOP-AUST, the CT angiography spot-sign was used to select patients who had evidence of ongoing haematoma expansion. Given the tighter recruitment time window and the additional time required to acquire and interpret CT angiography, we have not included this requirement in STOP-MSU. We would anticipate a high rate of haematoma growth in this patient group nonetheless based on the ultraearly time window.
STOP-MSU was ongoing at the beginning of the COVID-19 pandemic. In response to the pandemic, the trial executive committee carefully reviewed the trial protocol and implemented a number of statistical and practical strategies to mitigate the impact of the pandemic on the trial conduct and to ensure ongoing recruitment while maintaining participant safety and data validity.
The ultimate objective of this study is to investigate tranexamic acid as a potential therapeutic to acutely attenuate ICH growth. This is an inexpensive, safe and readily available drug, which can be easily delivered in a variety of settings including MSUs. Such a therapy could potentially have significant and widely generalisable implications for the treatment of this devastating condition.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
STOP-MSU was approved by the Human Research Ethics Committee of Melbourne Health (HREC/17/MH/142).
References
Footnotes
NY and HZ are joint first authors.
Twitter @#teddyyhwu, @AndrewKCheung
GAD and SMD as Joint Chairs of the Trial Executive Committee.
Contributors NY and HZ are the coprincipal investigators and trial coordinators. GAD and SMD are the trial co-chairs. LC is the trial biostatistician. TW and GS will perform imaging planimetric analysis. HM, BCVC, PJM, BY, MP and CL are members of the trial executive committee. All other authors are site principal investigators and have contributed to drafting and/or editing of this manuscript.
Funding Medical Research Future Fund, Australia (APP1152282).
Competing interests Henry Zhao has received grant funding from the Medical Research Future Fund (Commonwealth Government of Australia). Leonid Churilov is co-chairperson of the Australasian Stroke Trials Network (unpaid). Andrew Cheung has received honoraria for lectures/presentations from Medtronic and Stryker and support for attending meetings and/or travel from Stryker. Rohan Grimley is a member of the Clinical Council, Stroke Foundation (Australia). Chung Hsu has received grants from the Ministry of Health and Welfare in Taiwan. Peter Mitchell has received institutional research grants from Stryker and Medtronic, travel support for attendance of international conferences and honoraria for speaking at symposia from Stryker, and is an executive member and immediate past president of the Australian and New Zealand Society of Neuroradiology (ANZSNR).
Provenance and peer review Not commissioned; externally peer reviewed.