Article Text

Abstract
Background and purpose Stroke is the second leading cause of death worldwide and the leading cause of mortality and long-term disability in China, but its underlying risk genes and pathways are far from being comprehensively understood. We here describe the design and methods of whole genome sequencing (WGS) for 10 914 patients with acute ischaemic stroke or transient ischaemic attack from the Third China National Stroke Registry (CNSR-III).
Methods Baseline clinical characteristics of the included patients in this study were reported. DNA was extracted from white blood cells of participants. Libraries are constructed using qualified DNA, and WGS is conducted on BGISEQ-500 platform. The average depth is intended to be greater than 30× for each subject. Afterwards, Sentieon software is applied to process the sequencing data under the Genome Analysis Toolkit best practice guidance to call genotypes of single nucleotide variants (SNVs) and insertion-deletions. For each included subject, 21 fingerprint SNVs are genotyped by MassARRAY assays to verify that DNA sample and sequencing data originate from the same individual. The copy number variations and structural variations are also called for each patient. All of the genetic variants are annotated and predicted by bioinformatics software or by reviewing public databases.
Results The average age of the included 10 914 patients was 62.2±11.3 years, and 31.4% patients were women. Most of the baseline clinical characteristics of the 10 914 and the excluded patients were balanced.
Conclusions The WGS data together with abundant clinical and imaging data of CNSR-III could provide opportunity to elucidate the molecular mechanisms and discover novel therapeutic targets for stroke.
- stroke
- genetic
Data availability statement
Data are available upon reasonable request. Data in this article are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Stroke is the second leading cause of death worldwide, and the leading cause of mortality and long-term disability in China.1 Being the most common type of stroke, ischaemic stroke (IS) accounts for about 80% of all strokes,2 and more than 90% of IS are sporadic.3 IS is a complex multifactorial disease arising from complicated gene-environment interactions. Therefore, uncovering genetic contributions to IS could help to identify the genes, pathways and networks that are involved in IS pathogenesis. Although several novel genetic variants that were associated with IS susceptibility have been discovered in the last decades,4–9 few studies explored the correlation between genetic variants and stroke outcomes. Moreover, previous genetic studies on IS were mainly conducted in European and African populations,4 10 and there is limited data for the Chinese population. Due to the substantial ancestral difference,11 whether these reported IS-associated genetic variants could also contribute to IS pathogenesis in Chinese population needs verification.
The Third China National Stroke Registry (CNSR-III) is a nationwide prospective registry with 15 166 patients with IS or transient ischaemic attack (TIA) in China.12 A broad and comprehensive spectrum of individual-level data had been collected, including clinical phenotypes, aetiological classification, neuroimaging, biomarkers and clinical outcomes. The aetiological subtyping information was recorded centrally. Taking these advantages, we perform whole genome sequencing (WGS) for 10 914 patients in the prespecified genetic substudy of CNSR-III to delineate the genetic landscape of IS and TIA in Chinese population.
Methods
Patients
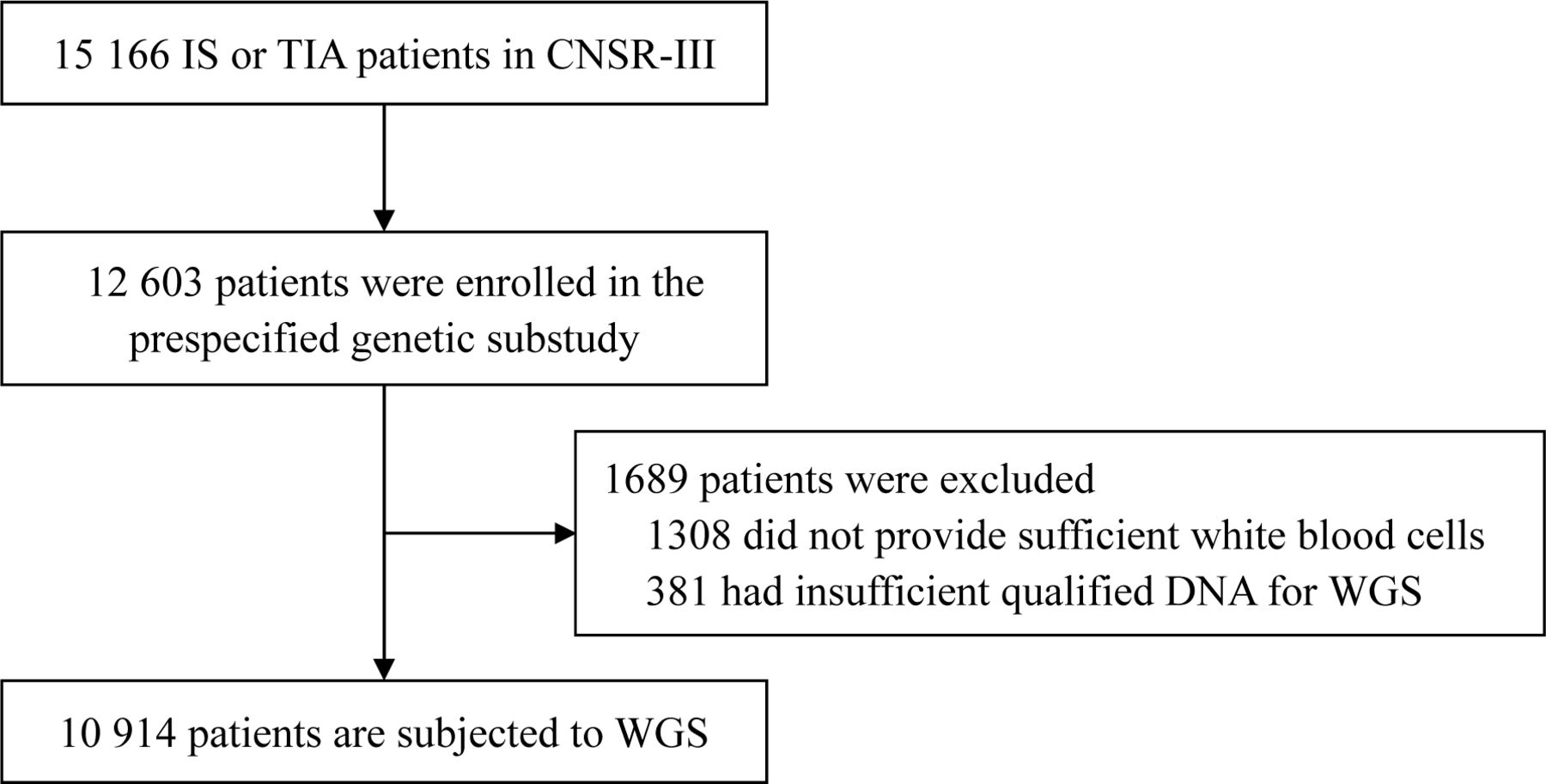
The CNSR-III is a nationwide prospective registry for patients presented to hospitals with acute ischaemic cerebrovascular events between August 2015 and March 2018 in China.12 There is a total of 15 166 patients with IS (n=14 146, 93.3%) or TIA (n=1020, 6.7%) within 7 days from the onset of symptoms to enrollment. The CNSR-III involved 201 hospitals that cover 22 provinces and 4 municipalities in China, including 163 grade III (central hospitals for certain district or city, usually teaching hospitals) and 38 grade II (hospitals serving several communities) urban hospitals. A total of 12 603 patients participated in the prespecified genetic substudy. The white blood cells (WBCs) from a total of 10 914 patients are applied in WGS (figure 1). The written informed consents were obtained from all patients or legally authorised representatives before entering into the study.

Flow chart of patient selection for WGS in the prespecified genetic substudy of CNSR-III. CNSR-III, The Third China National Stroke Registry; IS, ischaemic stroke; TIA, transient ischaemic attack; WGS, whole genome sequencing.
DNA extraction
For each sample, WBCs was used to extract the genomic DNA, which was performed using Magnetic Blood Genomic DNA Kit (DP329, TIANGEN Biotech Co Ltd, Beijing, China) on KingFisher Flex (Thermo Scientific Co, Massachusetts, USA) system for automatic genomic DNA extraction and purification at iGeneTech Co Ltd. (Beijing, China) or by manual phenol–chloroform DNA extraction at BGI Genomics (BGI-Shenzhen).
Evaluation of DNA quality
The concentration of genomic DNA was quantified using Qubit 2.0 fluorometer (Thermo Scientific Co, Massachusetts, USA) and SpectraMax Gemini XPS (Molecular Devices, San Francisco, USA) at BGI Genomics (BGI-Shenzhen). Electrophoresis was conducted on 1% agarose gel to make sure that the majority of genomic DNA segments was longer than 20 Kb and was not substantially degraded. Genomic DNA samples with concentration ≥12.5 ng/µL and total amount ≥0.5 µg was qualified for further procedures. For each of the qualified sample, the DNA is further applied in library construction and subsequent WGS process, as well as single nucleotide variant (SNV) genotyping (see details below).
Library construction
The qualified genomic DNA is randomly fragmented by ultrasound using CovarisLE220 (Covaris, Massachusetts, USA) according to the manufacturer’s instructions. The DNA fragments in the range of 200 to 400 bp are selected by VAHTSTM DNA Clean Beads (Vazyme Biotech Co, Ltd, Nanjing, China). The end repair for DNA fragments is performed by adding an ‘A’ nucleotide to the 3’ end of each strand. Afterwards, the dTTP-tailed adapters are ligated to both ends of the repaired/dA-tailed DNA fragments. The ligation product is then amplified by PCR. Then the products are purified by VAHTSTM DNA Clean Beads (Vazyme Biotech Co, Ltd, Nanjing, China). The purified PCR products with total mass ≥200 ng, and the main peak in 300 to 500 bp should be applied. Single strand separation is conducted by heat-denaturing the PCR product at 95 °C. Circularisation process is performed by mixing the single-stranded DNA fragments with splint oligos (sequence: GCCATGTCGTTCTGTGAGCCAAGG) and DNA Rapid Ligase to generate single-stranded DNA circles. The remaining linear molecule is digested with the exonuclease. The enzymatic digestion products are purified by Agencourt AMPure XP medium (Beckman Coulter, Indiana, USA). The single-stranded circle DNA (ssCir DNA) are formatted as the final library. The purified enzymatic digestion products are quantified with Qubit ssDNA Assay Kit (Thermo Scientific Co, Massachusetts, USA), and the final yield should be ≥12 ng.
BGISEQ-500 WGS sequencing
Rolling circle amplification is performed for the qualified libraries to produce DNA Nanoballs (DNBs). Then the DNBs are loaded into the patterned nanoarrays and 100 bp pair-end reads are sequenced on the BGISEQ-500 platform (BGI Genomics, Shenzhen, China). Sequencing-derived raw image files are processed by BGISEQ-500 base-calling software (V.1.2.1.21840) under default parameters settings. The sequence data are stored in FASTQ format. The average depth for each subject is intended to be greater than 30×.
SNV genotyping
To make sure that the DNA samples are neither mistaken nor contaminated during the WGS process, we selected 21 biallelic fingerprint SNVs and planned to genotype them for each participant of WGS. These 21 SNVs distribute on 15 different autosomes and are at least 13M apart. The minor allele frequencies of these SNVs are between 0.16 to 0.5 within the Han Chinese in Beijing samples in 1000 Genome Project.13 The SNV genotyping experiments are performed at BGI Genomics (BGI-Shenzhen) independently and simultaneously with WGS. For each sample, approximately 30 ng of qualified genomic DNA is used. Locus-specific PCR and detection primers are designed using the MassARRAY Assay Design software (Agena Bioscience, California, USA). Multiplex PCR and locus-specific single-nucleotide extension are performed for each DNA sample, then the products are desalted and transferred to a 384-well SpectroCHIP array. After MALDI-TOF (matrix-assisted laser desorption/ionization-time of flight) mass spectrometry, MassArray Typer software (V.4.1, Agena Bioscience, California, USA) is used to call the genotype for each participant.
After the accomplishment of WGS and SNV genotyping, the genotypes of the 21 SNVs are compared between those that are respectively obtained from WGS data analyses and MALDI-TOF mass spectrometry to verify that DNA sample and sequencing data originates from the same individual.
WGS data cleanup
Raw sequence reads are filtered using an in-house pipeline for quality control. The following steps are executed consecutively: Removing both of the paired reads if (1) any one of the reads contain sequencing adapter, (2) any one of the reads whose low-quality base ratio (base quality less than or equal to 12) is more than 50%, (3) any one of the reads whose unknown base (‘N’ base) ratio is more than 10%. Afterwards, fastp (V.0.20.0) is applied to filter out low-quality reads and bases,14 and downstream bioinformatics analyses are conducted on these qualified data.
Mapping and variant calling
The paired-end reads are processed under the Genome Analysis Toolkit (GATK) best practice guidance using Sentieon (release 201808.05, https://www.sentieon.com, bioRxiv 115717; doi:10.1101/115717).15 The reads are aligned to the hg38 human reference genome sequence that is downloaded from GATK bundle (ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/hg38/Homo_sapiens_assembly38.fasta.gz) using Burrows-Wheeler Alignment tool that is implemented in Sentieon. The SNVs and insertion-deletions (indels) in the regions of segmental duplications and unassigned chromosomes are ignored in the downstream analyses. For each sample, the base quality, sequencing depth, GC (guanine-cytosine) content, mapping rate, mismatch rate, duplication rate and coverage is calculated. After removing the duplicated reads and recalibrating the base quality scores, SNVs and indels are first called using Haplotyper of Sentieon for each individual and then jointly called for all of the participants. Then, variant quality score recalibration and hard filter methods are applied to obtain the high-quality variant calls for SNVs and indels. The ‘*.bam’ and ‘*.vcf’ files that are generated in the above procedures would be reserved for other researches. Copy number variations (CNVs) and structural variations (SVs) in the genome of patients are mainly called using GraphTyper2 and Manta.16 17
Population genetics analysis
To minimise problems arising from hidden family and population structure in the participants, we conduct the following quality control steps. First, kinship is explored by calculating pairwise identity-by-descent calculations for all pairs of individuals using PLINK (V.1.9).18 The existence of first and second degree relationships is checked using KING (V.2.1.8).19 Second, population structure is investigated using STRUCTURE software and by conducting principal component analysis.20 All of these analyses are conducted using autosomal SNVs and indels.
Variant annotation
Impact of the mutations on protein coding and protein truncating variants were predicted using variant effect predictor.21 Pathogenicity of SNVs and indels are evaluated using InterVar software (V.2.0.1) under guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.22 The potential impact of SNVs and indels on gene expression/regulation is investigated by reviewing GTEx, HaploReg and other databases or online tools.23 24 The impact of intronic and exonic mutations on pre-messenger RNA splicing is mainly predicted using SpliceAI.25
Biological significance of known or common CNVs and SVs are annotated by reviewing dbVar and Database of Genomic Variants.26 27 Novel CNVs and SVs are annotated by reviewing literatures on structure and function studies of the genes affected by the corresponding CNVs and SVs from PubMed.
Checking and reviewing
During the experimental procedures of this project, all of the WBCs and DNA loading, packaging, transferring and storing operations was conducted by one technician while being checked and supervised by another technician.
For WGS and SNV genotyping data, the MD5 code is generated for each data file before transfer, and is checked after the transfer. The commands and codes for WGS data mapping and variant calling are written by one bioinformatician while being reviewed by another bioinformatician. The log files are also reviewed and reserved.
All of the genetic information, clinical data and biospecimens are managed following the Regulations of the People’s Republic of China on Administration of Human Genetic Resources 2019.
Research projects
WGS data of 10K patients will be incorporated to identify the causality of certain risk factors for stroke outcomes, to investigate pleiotropic effect of genes on multiple phenotypes, and to understand the genetic relationship between particular comorbidities and IS. The accurate sequencing data from greater than 30× average depth in the WGS study also allows us to obtain a panoramic view of individual-specific variation and genetic structure of Chinese patients with IS or TIA. Some prespecified research topics are described below:
To draw a comprehensive genetic landscape of Chinese patients with IS or TIA, and characterise the geographical, lifestyle differences and their demographic origin;
To evaluate the genetic contribution to IS and its recurrent outcomes, especially the contribution of rare variants, CNVs and variants in certain region of the genome (eg, telomere and mitochondrial DNA);
To determine the causality of serum biomarkers for IS outcomes using association analyses and Mendelian randomisation;
To investigate the relationship between genetic features and brain imaging changes in IS;
To conduct the pharmacogenomics analyses on certain secondary prevention of IS;
To better understand the genetic mechanisms of IS with particular comorbidities (eg, chronic kidney disease, diabetes mellitus and hypertension).
Results
Among the 15 166 patients with IS or TIA in CNSR-III, 12 603 patients participated in the prespecified genetic substudy. Among them, 1308 participants did not provide enough WBCs. After DNA extraction and quality evaluation, the DNA of 381 participants was insufficient or unqualified. Therefore, a total of 1689 participants were excluded and WGS are conducted for 10 914 participants of CNSR-III (figure 1). The workflow of WGS and downstream bioinformatics analyses are shown in figure 2.

{kind=link}
{kind=link}
Workflow of WGS and bioinformatics analyses. The first two rows shows the process of DNA extraction, quality control, library construction and WGS. The third row demonstrates downstream bioinformatics analyses of WGS data. Some of the images are retrieved or adapted from Servier Medical Art (https://smart.servier.com/), which is licensed under a Creative Commons Attribution 3.0 Unported License. The photos of instruments are downloaded from websites of BGI Genomics (https://www.bgi.com/), Thermo Fisher (https://www.thermofisher.com/) and Agena Bioscience (https://agenabio.com/), respectively. DNB,DNA Nanoball; IS, ischaemic stroke; ssCir DNA, single-stranded circle DNA; TIA, transient ischaemic attack; WGS, whole genome sequencing.
Baseline clinical characteristics of the included 10 914 patients and excluded patients were presented in table 1. The average age was 62.2±11.3 years, and 31.4% of the patients were women. Patients diagnosed to be IS were 10 166 (93.2%), among which 50.4% had minor stroke (NIHSS (National Institutes of Health Stroke Scale score) ≤3). A total of 31.8% of the included patients were current smokers, and 14.5% were heavy drinkers (defined as ≥2 standard alcohol consumption per day). A total of 21.3% of the included patients had a history of IS. A total of 10.8%, 7.0% and 62.8% of the included patients had a history of coronary heart disease, atrial fibrillation and hypertension, respectively. The two groups of included and excluded patients were balanced regarding baseline characteristics (table 1).
Baseline characteristics of the included patients in the patients who underwent whole genome sequencing and the rest of the patients in CNSR-III
Discussion
Stroke is a complex disease that has multiple aetiologies. Genetic and genomic studies among populations from diverse ancestry could refine our understanding on molecular mechanism of stroke. Therefore, we conduct WGS for 10 914 patients from CNSR-III. The WGS procedures and baseline characteristics of patients are reported in this study. The WGS of CNSR-III constructs a genomic data set that facilitate large scale IS genetic analyses in Chinese population. The CNSR-III collected a comprehensive spectrum of phenotypic information under consistent and standardised criteria, which could increase the power and credibility of the genetic analyses. In addition, all of the patients are followed up for clinical outcomes,12 and this provides an opportunity for discovery of genetic variants that are associated with patients’ outcomes after stroke.
In contrast to DNA microarrays that were mainly used in previous genetic associations on IS,4 10WGS technology applied in this study could provide nearly all of the SNVs and indels, and simultaneously capture genetic information on CNVs and SVs for each patient. Therefore, WGS enables a systematic evaluation of the genetic effect of rare variants (allele frequencies <1% in population) to IS and TIA. As the contribution of the rare variants remains one of the top challenges in stroke genetics, the WGS study would provide a better understanding on IS and TIA pathophysiology.10 The average depth for WGS is intended to be greater than 30× in this project, because at this depth, both accurate variant calling and cost-effectiveness could be achieved.28 29 Moreover, >95% the genome could be covered by at least 10 sequencing reads, and >95% of the heterozygous variation could be accurately identified under this design.30 Therefore, the WGS could provide high-quality genetic data for further investigations on IS.
In conclusion, the WGS and genome-wide analyses on CNSR-III would help to refine our understanding on the genetic contribution to IS/TIA and stroke outcomes, and possibly discover novel therapeutic targets for secondary prevention.
Data availability statement
Data are available upon reasonable request. Data in this article are available upon reasonable request.
Ethics statements
Ethics approval
The study was approved by the ethics committees of Beijing Tiantan Hospital and all other research centres according to the principles expressed in the Declaration of Helsinki.
References
Footnotes
SC, ZX and YL are joint first authors.
Twitter @yilong
Contributors Study concept and design: SC, HaL and YoW. Drafting of the manuscript: SC, ZX and YL. Statistical analysis: AW, XH and ZX. Study supervision and organisation of the project: JL, YJ, XM, HaL, YiW and YoW. Supplying patients: ZW, GC, SW, ZJ, YC, XQ, JW, BS, WJ, ZA, WX, LZ, YG and HoL.
Funding This study was supported by grants from the Ministry of Science and Technology of the People’s Republic of China (2016YFC0901002, 2016YFC0901001), Beijing Municipal Science & Technology Commission (D171100003017002),Beijing Municipal Administration of Hospitals’ Mission Plan (SML20150502) and National Science and Technology Major Project (2017ZX09304018). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.