Article Text

Abstract
Different kinds of biological databases publicly available nowadays provide us a goldmine of multidiscipline big data. The Cancer Genome Atlas is a cancer database including detailed information of many patients with cancer. DrugBank is a database including detailed information of approved, investigational and withdrawn drugs, as well as other nutraceutical and metabolite structures. PubChem is a chemical compound database including all commercially available compounds as well as other synthesisable compounds. Protein Data Bank is a crystal structure database including X-ray, cryo-EM and nuclear magnetic resonance protein three-dimensional structures as well as their ligands. On the other hand, artificial intelligence (AI) is playing an important role in the drug discovery progress. The integration of such big data and AI is making a great difference in the discovery of novel targeted drug. In this review, we focus on the currently available advanced methods for the discovery of highly effective lead compounds with great absorption, distribution, metabolism, excretion and toxicity properties.
- artificial intelligence
- big data
- targeted drug
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Traditionally, the discovery of novel targeted drugs is an expensive long-term progress, costing billions of US dollars and more than 10 years. In the very beginning, a therapeutic drug target must be identified by traditional experimental methods. Then, structural biologists come to decipher the three-dimensional (3D) structures as well as their ligand-binding characteristics to reveal whether this is a druggable target. Subsequently, medicinal chemists and pharmacologists use high-throughput screening to find several highly effective lead compounds for further safety assessment as well as clinical trials. In general, the above procedures are costly and tedious. In November 2018, a study was conducted to estimate the total cost of trials for the development of novel Food and Drug Administration (FDA)-approved drugs. Surprisingly, the average cost of efficacy trials for the 59 new drugs approved by the FDA during 2015–2016 was $19 million.1 Therefore, it is necessary to overcome the limitations of the conventional drug discovery procedures by introducing efficient, low-cost and computational methods.
Compared with traditional drug discovery methods, rational drug design, mainly including computer-aided drug design (CADD), is more efficient and economical. Rational drug design integrates molecular docking to the ligand-binding pocket of a promising therapeutic target, computes the binding energy of each docked small molecule compound, and selectively chooses the best ones as candidates for subsequent experimental procedures. Today, there are more than 100 000 protein 3D structures deposited in Protein Data Bank (PDB) for molecular docking.2 In contrast to traditional methods, rational drug design has boosted the hit rate of drug screening by more than 100 times, from ~0.01% to 1%~2%. Moreover, CADD is a more multidiscipline method which integrates advanced bioinformatic techniques and sophisticated computational algorithms. Due to its relatively high hit rates, CADD method is becoming the fundamental basis of industrial drug discovery as well as academic research.3 Recently, artificial intelligence (AI) assisted drug discovery of me-better drug from hit discovery to animal tests within 46 days.4
Cancer-targeted drugs are the most successful drugs for the last three decades, thanks to comprehensive omics databases of cancer research. A lot of cancer-related proteins have been identified as therapeutic targets by computational data mining of transcriptome data in databases such as The Cancer Genome Atlas (TCGA),5 The Human Protein Atlas(THPA)6 and so on. Unfortunately for other diseases, such as stroke, vascular-related diseases and other genetic diseases, there are no similar integrated omics databases to provide sufficient big data. However, there are increasingly more single cell transcriptome data of various diseases publicly available.7–10 Thus, such data will be precious goldmines in terms of the discovery of therapeutic targets for stroke, vascular-related diseases and other genetic diseases. Moreover, supercomputers are speeding up lead identification and evaluation. In this review, we provide an overview of how the integration of big data and AI could help us to discover new therapeutic targets and their targeted lead compounds, as well as their absorption, distribution, metabolism, excretion and toxicity (ADMET) properties.
Virtual screening to discover targeted lead compounds
Virtual screening technology is the core of CADD. Based on the 3D structure or the quantitative structure–activity relationship model of the target biomacromolecules, the theory of molecular biology and computer science and other related fields is used as a technical basis to select the compounds that meet the expectations from the known small-molecule databases. Then, one or more experimental methods are selected for targeted drug screening for specific diseases. In the pharmaceutical world, virtual screening is often considered as a top CADD tool to screen large chemical structural libraries and reduce them to a set of candidate compounds related to specific protein targets.11 At present, virtual screening has been regarded as a materialised tool, widely recognised in search for lead compounds and the enhancement of compound activity.12
The basic processes of virtual filtering mainly include the following:
Target selection: this is the first step in virtual screening, and this step is crucial. Small molecular compounds target four large molecules: proteins, polysaccharides, lipids and nucleic acids. Proteins such as enzymes, ion channels and GPCR(G Protein-coupled Receptor) are often preferred as potential drug targets because they are highly specific and less toxic, such as the discovery of heat shock protein (Hsp90) inhibitors,13 the discovery of a selective inhibitor of Aurora A,14 the discovery of TASK-3 (KCNK9) channel blockers,15 the virtual screening for GPCR drug screening16 and so on.
Prepare the compound database: before starting a new virtual screening, we need to collect all the compound structures for a specific drug target. In recent years, a number of compound databases have been developed which store not only the structure of the compound molecules, but also many chemical and biological information, such as ZINC,17 PubChem18 and others.
Docking software: currently popular molecular docking software are Dock, AutoDock, MolDock, Maestro and so on.19 These software are available for use and are easy to operate, but when the number of compounds involved in docking is too large (eg, 1 million), large-scale molecular docking methods and strategies need to be adopted. Linux-based virtual docking always plays an important role when we perform high-throughput docking.
Scoring system: molecular docking is a computational method that predicts the preferred position of a molecule (ligand) relative to a second molecule (receptor) when the two molecules combine to form a stable complex, and then predict the binding strength or binding affinity between the receptor and the ligand. There are two main types of docking: rigid docking and flexible docking.20 In rigid docking, the receptor and ligand are immobilised so that the bond angle and bond length are constant. This docking speed is very fast, but lacks practical application because flexible docking allows for conformational transformation. In flexible docking, the conformation of the ligand and acceptor can be converted at will during the calculation. This docking method requires relatively high computing power, but it can most accurately calculate the docking results and is suitable for the accurate investigation of the identification between molecules. Based on the position and binding energy, a docking score will be calculated.
Biological experiment verification: the candidate compounds of highest docking score are verified by both in vitro and in vivo biological experiments.
Clinical study: once all preclinical studies of these candidate compounds are proved to be effective, clinical studies will be performed on candidate compounds to determine their safety and effectiveness on patients.
Identification of ligand-binding pocket on the 3D protein model
The interaction between protein and ligand usually occurs in a pocket formed by conserved amino acids. The protein function relies on the ligand-binding site on its 3D structure. The identification of the binding pocket helps to discover new drugs and better understand the mechanism of actions of drugs, such as the discovery of a conservative pocket of the guanylate cyclase heme domain.21 In the general molecular docking calculation, an indispensable step is to define the binding position of the ligand molecule, that is, its binding pocket. If the binding site is known, the ligand type and protein function can be determined by computer and experimental procedures, and can be used in drug design and to predict potential side effects.22
Bioinformatics is a cross-disciplinary discipline that solves biological problems through the use of computer, mathematical and statistical methods. The determination of binding pockets is very important for designing drugs. Traditional X-ray crystallography and nuclear magnetic resonance methods predict large amounts of protein structures that are time-consuming and expensive, but bioinformatics provides different tools to predict the 3D structure of proteins and reveal their binding regions. Its application is very promising, such as the identification of conserved binding pockets in ricin A chain,23 RASSF2 potential binding pocket prediction24 and so on. There are two ways to find a pocket combination: (1) proteins with known 3D structures can be searched from the PDB database,25 and related information can be downloaded directly from the database; and (2) method of homology modelling, using I-TASSER, SwissModel, ModWeb and other online servers based on homologous modelling to generate protein 3D structure, as well as to predict the ligand-binding pocket, for example, prediction of serotonin 1A receptor binding pocket.26
Discovery of targeted lead compounds for a novel drug target
The drug target is a special site formed by biomolecules, and the drug can be combined with it to produce pharmacological effects (targeted agonist/inhibitor) for the purpose of preventing and treating diseases. According to the biological characteristics of biomolecules, drug targets can be classified into receptors, enzymes, ion channels, DNA, hormones and growth factors. The research and development (R&D) of new drug is a work with high investment and low yield. The discovery and confirmation of drug target is the first step of the R&D of a new drug. However, the number of clinically validated drug targets is still very small, so there is an urgent need to discover more new drug targets.
With the development of life science and bioinformatics, more and more target structures have been analysed. Different from traditional drug research methods, big data mining is widely used in drug target research, such as using genetic algorithm and bagging-svm ensemble classifier to predict drug targets,27 mining and forecasting cancer-related database,28 and using genetic disease-related data to predict novel therapeutic targets by computational data mining methods.29
The human genome database shows that there are more than 20 000 proteins in the human body, while the DrugBank database indicates only about 500 have been identified in the past 100 years.30 Therefore, there are many potential targets to be discovered and confirmed. Thanks to structure biologists, a lot of new biological processes mediated by protein–protein interaction, protein–DNA interaction and protein–RNA interaction have been discovered. These above proteins may probably serve as potential novel drug targets in the near future. The information of the drug target database can be used to analyse the sequence characteristics and biochemical characteristics of structural features, and to establish a prediction model to discover new drug targets. Therefore, we set up a set of novel methods for potential cancer-related drug target discovery, such as the following procedures: (1) TCGA and Human Protein Atlas databases were used to mine the data of targets related to prediction of cancer prognosis in the database. (2) Then use the computer to correlate with known cancer prognosis-related targets and score according to the correlation strength. (3) Then review the research progress of the target according to the score table and explore the 3D structural information of the drug target in the PDB database. (4) According to the integrated information, select the appropriate targets for further biological verification.
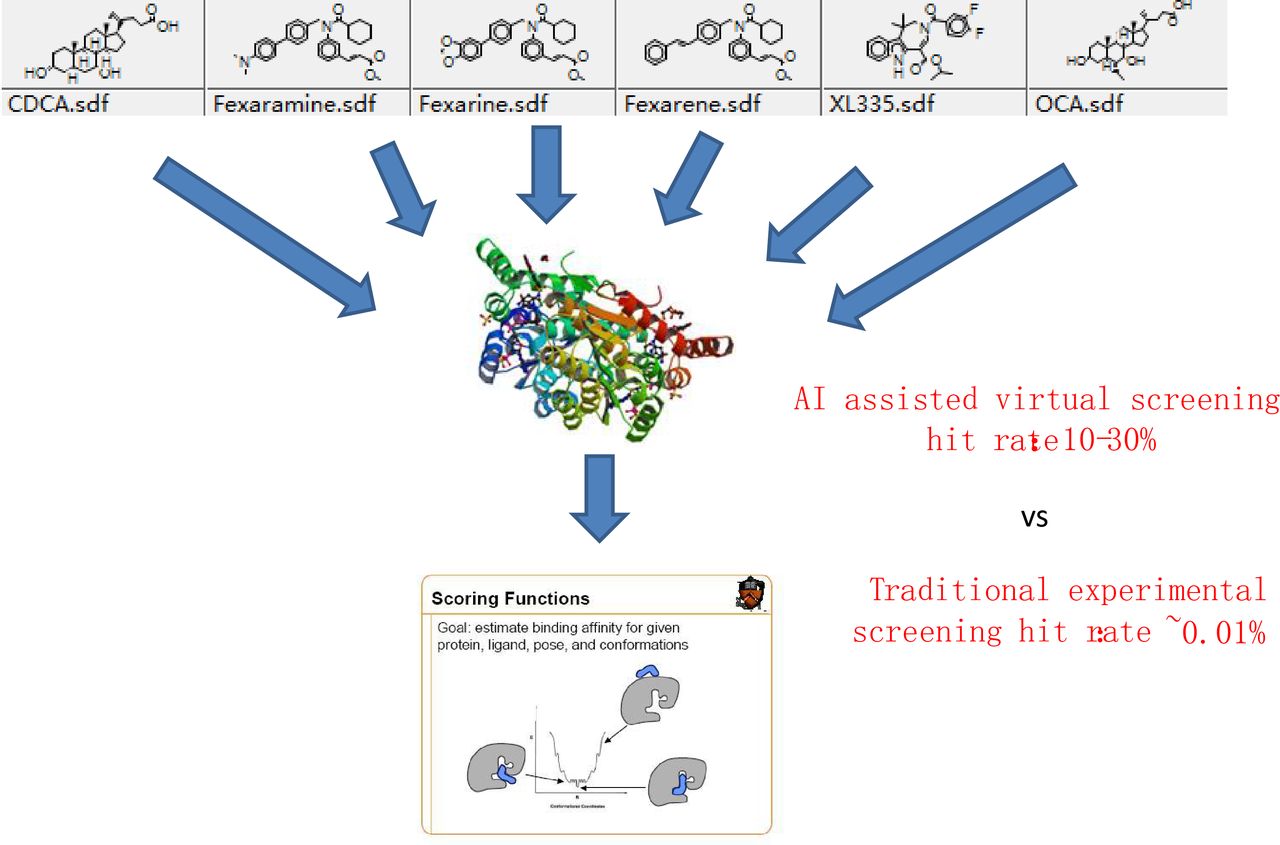
After successful verification of the novel therapeutic targets in vitro and in vivo, the virtual screening molecular docking-based drug screening can be performed according to the novel targets. This process has greatly reduced the time and cost compared with traditional drug development. In the past few years, our lab has discovered 73 novel compounds as well as 12 FDA-approved drugs targeting more than 30 potential novel therapeutic targets (figure 1). Moreover, four FDA-approved drugs will be used for clinical trial tests to cure multiple sclerosis in the near future (Drug repositioning, unpublished data,Jingwei Jiang).

Schematic procedure of artificial intelligence (AI)-assisted virtual screening. Millions of structurally diverse chemical compounds are docked to a specific therapeutic target. AI scoring function is used to select the best hits from millions of docked results.
Reverse docking to find drug targets of an old drug
Drug repositioning, also known as drug repurposing, defines new indications for existing drugs and can be used as an alternative to drug development.31 The benefits of repositioning include the availability of chemical materials and previously generated data that can be used, so the potential for R&D is significantly greater than the time and cost-effectiveness of bringing new drugs to market. It has been reported that there have been identified 109 molecules with other activities through in vitro screening, with these products having at least one marketing approval for a common disease indication or one marketing approval for a rare disease from the FDA’s rare disease research database.32 In our meta-analysis, a study shows that the class III antiarrhythmic amiodarone was active in neurodegeneration assays and could also selectively remove embryonic stem cells, and that the antipsychotic trifluoperazine was active in neurodegeneration assays.32 In contrast to traditional molecular docking, reverse docking is used for identifying receptors for a given ligand among a large number of crystal structures. It can be used to discover new targets for existing drugs and natural compounds, alternative indications of drugs through drug repositioning, and detecting adverse drug reactions and drug toxicity.33 Generally, the following steps are required to perform a drug repositioning by reverse docking (drug repositioning): (1) data set collection; (2) data set partition; (3) molecular descriptor calculation and modelling; (4) ensemble learning; (5) retrospective screening campaigns; (6) building positivity predictive value surfaces and choosing an adequate score threshold value; (7) prospective virtual screening; (8) molecular docking; and (9) reverse docking scoring. The results of the reverse docking were then verified by biological experiments. There are reports that they have implemented a computer-aided drug repurposing campaign to discover new inhibitors of falcipain-2. Four hits were acquired and tested against the enzyme, with two of them confirming inhibitory activity.34 The abandoned drug odanacatib displayed competitive inhibition, while the antibiotic methacycline also showed inhibitory effects through non-competitive inhibition.34 Therefore, it is feasible to find the target of the old drug through reverse docking. This method saves a lot of time and can reduce many experimental costs and experimental steps.
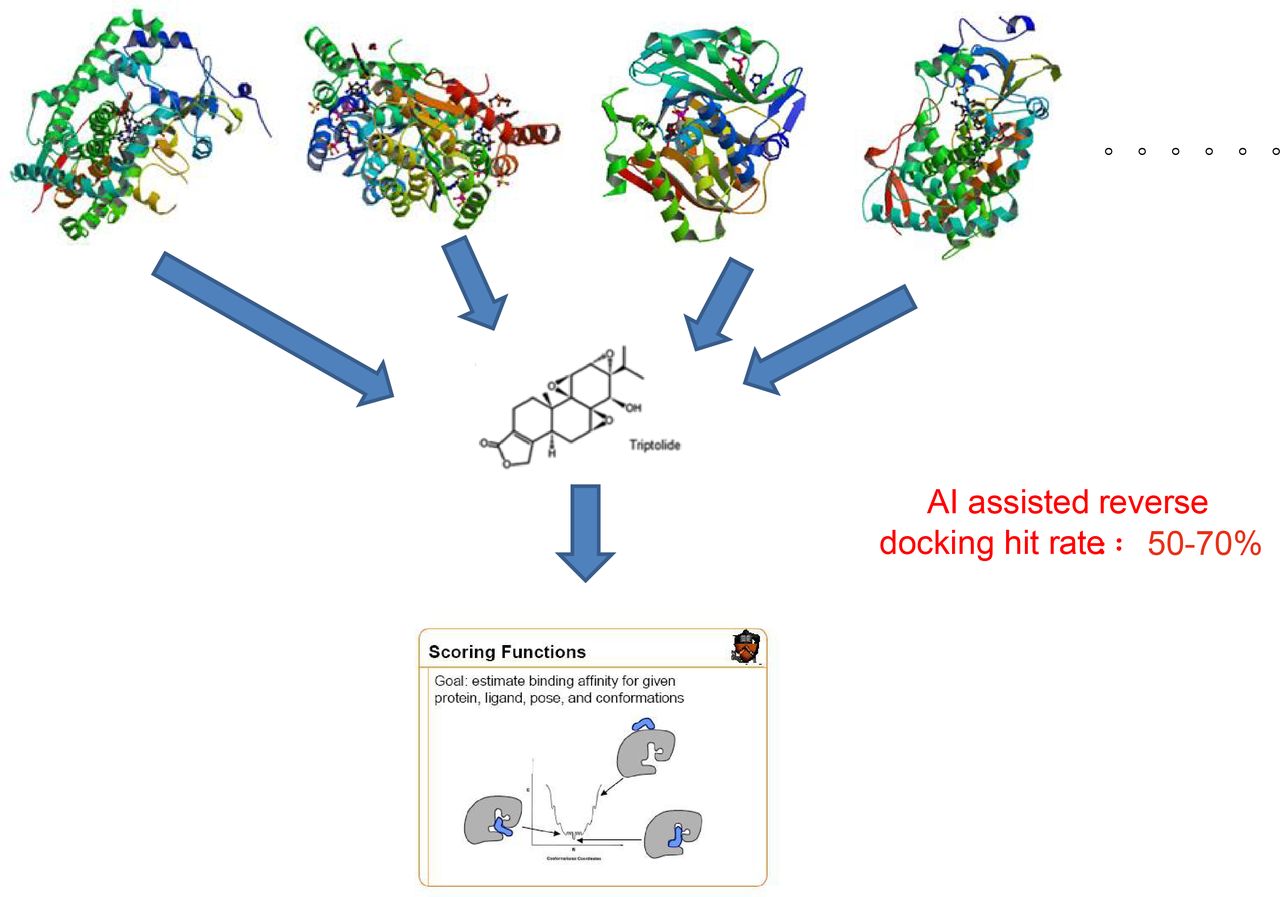
In the past few years, our lab has discovered 13 new targets for eight FDA-approved drugs through reverse-docking the old drugs to all ligand-binded structures extracted from PDB (>100 000 proteins; figure 2). The new indications and adverse effects of these old drugs have been revealed through biological verification for those reverse-docked targets (unpublished data,Jingwei Jiang).

Schematic procedure of artificial intelligence (AI)-assisted reverse docking. More than 100 000 structurally diverse protein structures are reversely docked to a specific chemical compound/natural product. AI scoring function is used to select the best hits from millions of docked results.
AI for the prediction of a compound’s ADMET
The ADMET of chemicals plays a key role in drug discovery and development. High-quality drug candidates should not only have sufficient efficacy for the treatment target, but should also display appropriate ADMET characteristics at the therapeutic dose.35 36 Moreover, ADMET’s predictions not only reduce the risk of late-stage attrition of new compounds and compound libraries but also help researchers optimise screening and testing by looking at only the most promising compounds.37 Just relying on biological experiments to verify the ADMET of a compound is a waste not only of time but also a lot of human and material resources. With the increase in computer speed and the implementation of quantum chemistry methodology, pharmacodynamic and pharmacokinetic issues have become computationally easier to handle. Quantum mechanics provides pharmaceutical scientists with the opportunity to study pharmacokinetic problems at the molecular level prior to laboratory preparation and testing.38 In order to realise ADMET for predicting compounds by computer, we need to do a lot of work in the early stage: (1) data collection and preparation (this is a crucial step); (2) calculation of ADMET-related properties based on the collected data; (3) definition of the ADMET score; and (4) validation of the ADMET score.36 An article on predicting the antimalarial activity of artemisinin derivatives showed that their predicted results showed significant antimalarial activity of compounds A24, A24a, A53, A54, A62 and A64. Subsequent studies of the derivative A64 showed that the experimental results of the derivative were well agreed with the predicted values.39 Although it is not guaranteed that the predicted results are completely consistent with the later experimental results, the introduction of AI can reduce many unnecessary troubles for later research. Machine learning (including AI) methods are accompanied by verification procedures in many cases and are often used in conjunction with other methods. Therefore, this makes them an excellent and attractive hybrid tool for reducing false predictions and model errors.40
In the past few years, our lab has developed several new ADMET prediction tools based on deep learning AI, such as prediction for logBB and logPapp to calculate the overall ADMET properties of a specific compound (figure 3A,B). Toxicity of a compound is very difficult to predict, mainly because it depends not only on its own chemical structure but also its direct actions on the target proteins. Hence, we collected all FDA-approved/withdrawn drugs (June 2019) to perform batch reverse docking with all ligand-binded structures extracted from PDB. Every docked target of each drug was scored and each drug can be considered as an N-dimensional vector in an N-dimensional space (figure 3C,D). Therefore, FDA-approved/withdrawn drugs can be referred as training data set to predict the toxicity of any given compounds (unpublished data,Jingwei Jiang).

{kind=link}
{kind=link}
{kind=link}
AI-assisted ADMET properties prediction. (A) Deep learning algorithm to calculate logBB for a specific chemical compound. (B) Deep learning algorithm to calculate logPapp for a specific chemical compound. (C) PCA(Principal Component Analysis) analysis on 48 186 reverse-docked proteins for 55 FDA-approved drugs (yellow dots) and 224 FDA-withdrawn drugs (blue dots). (D) PLS-DA(Partial Least Squares Discriminant Analysis) analysis on 48 186 reverse-docked proteins for 55 FDA-approved drugs (blue dots) and 224 FDA-withdrawn drugs (yellow dots). ADMET, absorption, distribution, metabolism, excretion and toxicity; AI, artificial intelligence; FDA, Food and Drug Administration;TPSA,total polar surface area.
Mining cancer database to discover novel therapeutic drug targets
Targeted drug design has become a hot topic because it is one of the key technologies for the discovery of therapeutic drugs. However, it is very difficult to find new drug targets through traditional experiments and methods and it is often difficult to achieve the desired results. Therefore, bioinformatic technology can be used to discover and identify new drug targets by mining cancer database. With the complete information of cancer genome/transcriptome sequencing accumulated in recent years, a variety of publicly available biological databases have provided us with a multidisciplinary goldmine of big data; especially the cancer genomic/transcriptomic/proteomic research has taken a big step forward.
TCGA is a project jointly supervised by the National Cancer Institute and the National Human Genome Research Institute. It aims to use high-throughput genome analysis technology to help people to better understand the occurrence and development of cancer, in order to achieve the purpose of prevention, diagnosis and treatment.41 For example, as of 2012, the genomes and epigenetic groups of lung squamous cell carcinoma have not been fully elucidated, but through the genomic and epigenetic analyses of about 180 lung SQCCs (Squamous Cell Carcinoma), TCGA has successfully screened out molecular targeting drugs for SQCC.42 Similarly, for ovarian serous cystadenocarcinoma, which is not optimistic in diagnosis and treatment at present, some potential therapeutic targets have been found through the comprehensive analysis of ovarian serous cystadenocarcinoma with higher grade by TCGA.43 In this way, the mining of the cancer database plays an important role in finding new therapeutic druggable targets.
By mining TCGA and THPA, our lab has discovered more than 10 potential novel therapeutic targets in various cancers, such as pancreatic cancer, lung cancer, triple negative breast cancer, colorectal cancer and so on, as well as their targeted compounds recently (unpublished data,Jingwei Jiang). For other diseases (such as stroke, cardiovascular diseases, neurological diseases and so on), there is no such intact database for the data mining to discover novel therapeutic targets. However, single cell transcriptomic sequencing data have been accumulated rapidly in the recent years, and these data will be helpful for new therapeutic target discovery in the near future.
8.Animal models and their limitations
In order to study the physiological and biochemical processes of human diseases and to explore the pharmacodynamics and pharmacokinetics of drugs in vivo, many animal models of various diseases have been introduced to preclinical studies. The most popular animal models are mouse, rat and monkey. Particularly, specific genes knock-out/knock-in mouse models have revolutionised our ability to study specific gene and protein functions in vivo and to better understand their molecular pathways and mechanisms.44
Although there are animal models used as powerful support for modern medical research in preclinical studies of many diseases, the new drug therapy is still difficult to convert from laboratory to clinical, because it is not feasible to mimic all aspects of a human disease in an animal model, especially a heterogeneous disease with complex pathophysiology such as stroke, and most of its studies are carried out in young animals without any complications. These models are physiologically different from real stroke, which especially affects the elderly with a variety of cerebrovascular risk factors.45 Therefore, in stroke studies, more than 1000 drugs were candidates in stroke models, but only 17 were tested in humans.46 Recently, many Alzheimer’s disease (AD) candidate drugs have shown great effects in mouse models but all failed during clinical trials. Perhaps this is because the tissues, organs and systems of animals are always different from those of human beings, and their reactions and effects to drugs are also different. An animal model cannot involve all aspects of a human disease. The age, sex and species of animals, tissue and organ damage, or the increase, deletion and change of genes caused by the establishment of animal models may have a significant impact on the experimental results.
Furthermore, another big problem of an animal model is the genetic difference between the animal protein and human protein. According to ENSEMBL genome database, orthologous genes have been analysed in human, chimpanzee, mouse and rat. Surprisingly, there are only 7043 orthologous genes (single copy common genes) shared in these four species. For chimpanzee and human, a set of 13 454 pairs of human and chimpanzee genes with unambiguous 1:1 orthology have been identified. Orthologous proteins in human and chimpanzee are extremely similar, with ~29% being identical and the typical orthologue differing by only two amino acids, one per lineage.47 Compared with ~25 000 genes in each of these four species, 7043 orthologous genes are ~28%, which means the other ~72% expressed non-orthologous proteins in these four species are very different in their protein sequences. Even if humans and chimpanzees are considered as the closest primate relatives in the animal kingdom, only 13 454 pairs of orthologue genes are identified consisting ~50% of their own expressed genes, which means the other ~50% expressed non-orthologous proteins are very different in their amino acid sequences. Taken together this above genetic evidence, it is very clear that if the drug target of the animal model is structurally different from the one of human, drugs targeting the animal protein will perform a significantly different effect between animal experiment and clinical experiment. The interaction between drug and its target is caused by hydrogen bonds, Van der Waals force and π-π interaction, which are exerting their interactive forces within less than 4 Angstrom. One or two amino acid mutations within the binding pocket of the drug target can make a big difference.
Cancer-targeted drugs are much more successful compared with targeted drugs developing for stroke and AD. Perhaps there are two major reasons. First, in the field of cancer-targeted drug R&D, there are a lot of mouse models carrying humanised genes (such as mouse carrying humanised immune system) to mimic the human immunity system. Second, patient-derived xenograft models (mouse carrying clinical human cancer tissue) have been widely introduced in the preclinical studies of cancer-targeted drugs. For stroke, AD and rare diseases, similar humanised animal models carrying human drug target protein must also be introduced in the preclinical studies in the near future.
Conclusion remark
Today, big data and AI are developing so fast that boost targeted drug discovery in an unprecedented speed. With the integration of various disease databases, scientists are able to perform data mining for de novo therapeutic target discovery. With AI assistance, novel identified therapeutic targets can be virtually screened for the discovery of targeted old drugs/new compounds within very short period. With AI-assisted reverse docking, old drugs or natural products could be repurposed for new indications very efficiently. ADMET properties can also be predicted by AI deep learning models to boost the success rate of in vivo experiments. Finally, with the help of 3D therapeutic target structural alignment, scientists can identify the difference on the drug binding pocket of a specific therapeutic target between human and animal models, and the selection of animal model must be considered very carefully in terms of their target 3D similarity.
References
Footnotes
Contributors All authors wrote the manuscript. JJ provided guidance and modifications.
Funding This work was supported by NSFC (no 81872892 and no 2018ZX09735001-004) and 'Double First Class' University project (no CPU2018GY20 and no CPU2018GY38).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.