Article Text

Abstract
Objectives The predisposition of intracranial atherosclerotic disease (ICAD) to East Asians over Caucasians infers a genetic basis which, however, remains largely unknown. Higher prevalence of vascular risk factors (VRFs) in Chinese over Caucasian patients who had a stroke, and shared risk factors of ICAD with other stroke subtypes indicate genes related to VRFs and/or other stroke subtypes may also contribute to ICAD.
Methods Unrelated symptomatic patients with ICAD were recruited for genome sequencing (GS, 60-fold). Rare and potentially deleterious single-nucleotide variants (SNVs) and small insertions/deletions (InDels) were detected in genome-wide and correlated to genes related to VRFs and/or other stroke subtypes. Rare aneuploidies, copy number variants (CNVs) and chromosomal structural rearrangements were also investigated. Lastly, candidate genes were used for pathway and gene ontology enrichment analysis.
Results Among 92 patients (mean age at stroke onset 61.0±9.3 years), GS identified likely ICAD-associated rare genomic variants in 54.3% (50/92) of patients. Forty-eight patients (52.2%, 48/92) had 59 rare SNVs/InDels reported or predicted to be deleterious in genes related to VRFs and/or other stroke subtypes. None of the 59 rare variants were identified in local subjects without ICAD (n=126). 31 SNVs/InDels were related to conventional VRFs, and 28 were discovered in genes related to other stroke subtypes. Our study also showed that rare CNVs (n=7) and structural rearrangement (a balanced translocation) were potentially related to ICAD in 8.7% (8/92) of patients. Lastly, candidate genes were significantly enriched in pathways related to lipoprotein metabolism and cellular lipid catabolic process.
Conclusions Our GS study suggests a role of rare genomic variants with various variant types contributing to the development of ICAD in Chinese patients.
- genetics
- stroke
Data availability statement
Data are available upon reasonable request. Anonymised data can be available for qualified investigators upon request to the corresponding authors.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
As a global major ischaemic stroke subtype, intracranial atherosclerotic disease (ICAD) constitutes 33%–50% of all ischaemic strokes in East Asians, compared with 5%–10% in Caucasians.1 The predisposition of East Asian populations to ICAD might suggest a genetic basis, which remains largely unknown.
As a multifactorial disease attributable to inheritable, environmental and pathophysiological factors, the genetic aetiology of ICAD is still elusive.2 Several genome-wide association studies (GWAS) have been conducted to identify the associations between common single-nucleotide polymorphisms (SNPs) and ICAD; however, most of these SNPs result in very small effect sizes,3 and no causitive genes or variants to ICAD with solid evidence provided are currently available. Instead, monogenic traits have been demonstrated in patients with conventional vascular risk factors (VRFs), an intermediary status beyond ICAD, or in cases with other stroke subtypes.3 First of all, VRFs, such as hypertension, dyslipidaemia and diabetes, are well-established modifiable risk factors for ICAD.4 Our previous study suggests a significantly higher prevalence of conventional VRFs and intracranial stenosis in Chinese patients with minor ischaemic stroke or transient ischaemic attack (TIA), compared with contemporarily recruited Caucasian counterparts.5 It implies that the difference in genetic background contributing to VRFs across different populations could underlie the differences in the number of cases of ICAD seen in various ethnicities. In addition, in patients who had a stroke, ICAD can coexist with other stroke aetiologies. For example, 51% and 85% of patients who had a stroke with ICAD were reported to have coexisting cardioembolic pathology and small vessel disease, respectively.6 It suggests ICAD and other stroke subtypes might share genetic factors. Thus, genetic variants involving genes related to VRFs and/or other stroke subtypes may also contribute to ICAD but have not been well elaborated.
Recently, several studies using exome sequencing in small cohorts (less than 30 samples/families) suggested that rare genomic variants could contribute to stroke occurrence,7 8 while there was no dedicated study in a pure phenotype of ICAD-related stroke (ie, symptomatic ICAD). With an objective to investigate the potential genetic basis of symptomatic ICAD, we performed genome sequencing (GS, 60-fold) with well-established pipelines in a well-characterised cohort of Chinese symptomatic patients with ICAD to detect rare genomic variants in genes potentially related to ICAD.
Materials and methods
Patient recruitment
As described in our previous study,9 we prospectively recruited adult Chinese patients of Han ethnicity with acute ischaemic stroke attributed to high-grade ICAD (60%–99% stenosis) confirmed in magnetic resonance angiography (MRA), CT angiography (CTA) and/or digital subtraction angiography (DSA). The severity of luminal stenosis in symptomatic ICAD was measured by the Warfarin–Aspirin Symptomatic Intracranial Disease criteria,10 in DSA if available, otherwise in CTA/MRA. The infarct topography in a stroke protocol by 3 T magnetic resonance (MR) examination needed to be concordant with ICAD. Stroke aetiology and relevance to ICAD were determined by experienced neurologists, based on clinical syndrome, imaging features and concurrent cardiovascular risk factors. Commonly seen infarct topography in symptomatic ICAD was previously described.11 We excluded patients with probable non-atherosclerotic stenosis (eg, moyamoya disease, dissection or infective/autoimmune vasculitis), evidence of cardioembolism (eg, atrial fibrillation, valvular heart disease or myocardial infarction within 6 weeks), concurrent tandem of >70% extracranial carotid or vertebral artery stenosis, and contraindications to MR examination or contrast angiography. We recorded demographics, status of conventional VRFs (ie, hypertension, dyslipidaemia and diabetes; a known history on admission of the index stroke or newly diagnosed on discharge) of all participants at baseline. A total of 126 Chinese subjects from Hong Kong (50% men) without ICAD (age at testing 37.0±6.3 years) were also recruited for GS as a representative of the general Hong Kong Chinese population for variant frequency estimation. After obtaining a written informed consent, 2 mL peripheral blood was taken from each patient in an EDTA tube.
Genome sequencing
DNA was extracted with DNeasy Blood and Tissue Kit (cat number/ID: 69506; Qiagen, Hilden, Germany), fragmented (~450 bp) and subjected to library construction with KAPA Library preparation kit (Kapa Biosystems, Wilmington, Massachusetts, USA). After end repair, A-tailing and adapter ligation, PCR amplification was performed. The concentration of each library was measured by Qubit (Thermo Fisher, Waltham, Massachusetts, USA) and subjected for GS at a minimal read-depth of 60-fold, paired-end 150 bp on a HiSeq X Ten platform (Illumina, San Diego, California, USA).
Genomic variant detection
After quality control, paired-end reads were aligned to the human reference genome (GRCh37/hg19) by Burrows-Wheeler aligner and then reformatted by SAMtools. Detection of single-nucleotide variant (SNV) and small insertion/deletion (InDel), was performed by HaplotypeCaller V.3.4 of the Genome Analysis Toolkit (Broad Institute), and annotation was conducted by ANNOVAR12 with public databases and in-house datasets.13
The analysis of aneuploidy, copy number variant (CNV) and structural rearrangement were previously described.14 15 Aneuploidy is defined as whole chromosome gained or lost. CNV analysis was at a resolution of 50 kb. Uniquely aligned reads were classified into adjustable sliding windows (50 kb with 5 kb increment) for detecting the candidate region(s) with CNVs, and reads were also classified into non-overlapping windows (5 kb) for identifying the precise boundaries of CNV(s). All chimeric read pairs, of which each end was aligned to different chromosomes or to the same chromosome but with a distance larger than the expected insert size (>10 kb), were collected for identifying chromosomal structural rearrangements. After clustering of chimeric read pairs, those events that potentially resulted from systematic errors/random errors or with incorrected aligned orientations were filtered out. Mosaicism refers to the variants presented in part of the cells, while constitutional event is defined as the variants identified in all cells.
Variant analysis and interpretation
We performed a literature review and identified 155 genes in monogenic traits (online supplemental eTable 1), including (1) 50 genes related to conventional VRFs (ie, hypertension, diabetes and dyslipidaemia); and (2) 105 genes related to other stroke subtypes (eg, large artery atherosclerotic stroke, moyamoya disease, small vessel stroke (SVS) and cardioembolic stroke).
Supplemental material
Potential deleterious SNVs/InDels were selected based on the following criteria: (1) read-depth of ≥10, mutant allele fraction of ≥30% and ≤70% for heterozygous variants; >70% for homozygous variants; (2) affecting protein-coding region and/or exon-intron junctions±10 bp; (3) with a minor allele frequency (MAF) of ≤1% in East Asians indicated in the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org) according to previous publications in adult disease studies;7 16 (4) variants predicted as ‘pathogenic’, ‘likely pathogenic’ or ‘uncertain significance’ by InterVar (http://wintervar.wglab.org). Variants reported as pathogenic or likely pathogenic in ClinVar or ‘disease-causing mutation’ or ‘likely disease-causing mutation’ in the Human Gene Mutation Database (HGMD) were classified as known variants. Variants not reported in ClinVar or HGMD but (1) predicted as ‘deleterious’ by at least two in silico predictions such as SIFT, Polyphen2 HumVar, Mutation Taster and CADD (≥20); (2) likely resulting in alternative splicing sites predicted by Human Splicing Finder; or (3) as truncating mutations in genes with a pLI (the probability of being loss-of-function intolerant) score of ≥0.9 were also selected. CNVs identified as common CNVs in the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home) or gnomAD (gnomAD V.2.1.1), or occurring at a frequency of >1% in our in-house database were filtered out as polymorphisms. Known pathogenic/likely pathogenic CNVs or rare CNVs involving genes from the aforementioned gene lists were selected for further analysis. We further analysed chromosomal structural rearrangement results in potential gene dosage effect, direct disruption or potential gene dysregulation by disruption of interaction between regulatory elements and gene(s) or topological associated domains in relation to ICAD. Candidate variants were validated with alternative methods such as Sanger sequencing, array-based comparative genomic hybridisation (aCGH) chromosomal microarray analysis (CMA) and karyotyping we previously reported(online supplemental eTable 2).14 15
Retrieval of MAF
MAFs of each SNV/InDel were enquired in local non-ICAD Chinese subjects sequenced by GS (n=126), general Chinese individuals (Nyuwa Chinese Population Variant Database (NCVD), n=2999; http://bigdata.ibp.ac.cn/NyuWa_variants/) and Chinese subjects with metabolic traits and diseases (ChinaMAP, n=10 588; http://www.mbiobank.com/), which are probably at a higher risk of developing ICAD.17 In addition, MAFs were also enquired in public databases of the general populations in Europe and East Asia (gnomAD .2.1.1).
Gene enrichment analysis
Gene enrichment analysis for pathway or gene ontology was performed with Metascape18 (https://metascape.org/gp/index.html#/main/step1) by providing the gene list, and log10(q) <−2 was regarded as statistical significance.
Data availability
Anonymised data can be available for qualified investigators on request to the corresponding authors.
Results
Overall, among 152 unrelated symptomatic patients with ICAD strictly phenotyped by clinical information and imaging exams from 2007 to 2016, 92 patients (71.7% men) who consented to further GS investigation of the underlying genetic aetiology were included in the current analysis. The mean age at stroke onset was 61.0±9.3 years; 76.1%, 85.9% and 40.2%, respectively, had a known history of or newly diagnosed hypertension, dyslipidaemia and diabetes on discharge. The culprit ICAD lesion was in terminal internal carotid artery, middle cerebral artery, terminal vertebral artery or basilar artery in 10 (10.9%), 72 (78.3%), 3 (3.3%) and 7 (7.6%) patients, respectively. The mean luminal stenosis in the symptomatic ICAD lesions was 78.6%±8.6%, measured in DSA in 87 patients and CTA/MRA in 5 patients. We performed GS (with an average coverage of 62.3-folds) in each of these 92 patients to detect the rare and potentially deleterious genomic variants in genes related to VRFs and/or stroke subtypes.
Rare and potentially deleterious SNVs/InDels identified in symptomatic patients with ICAD
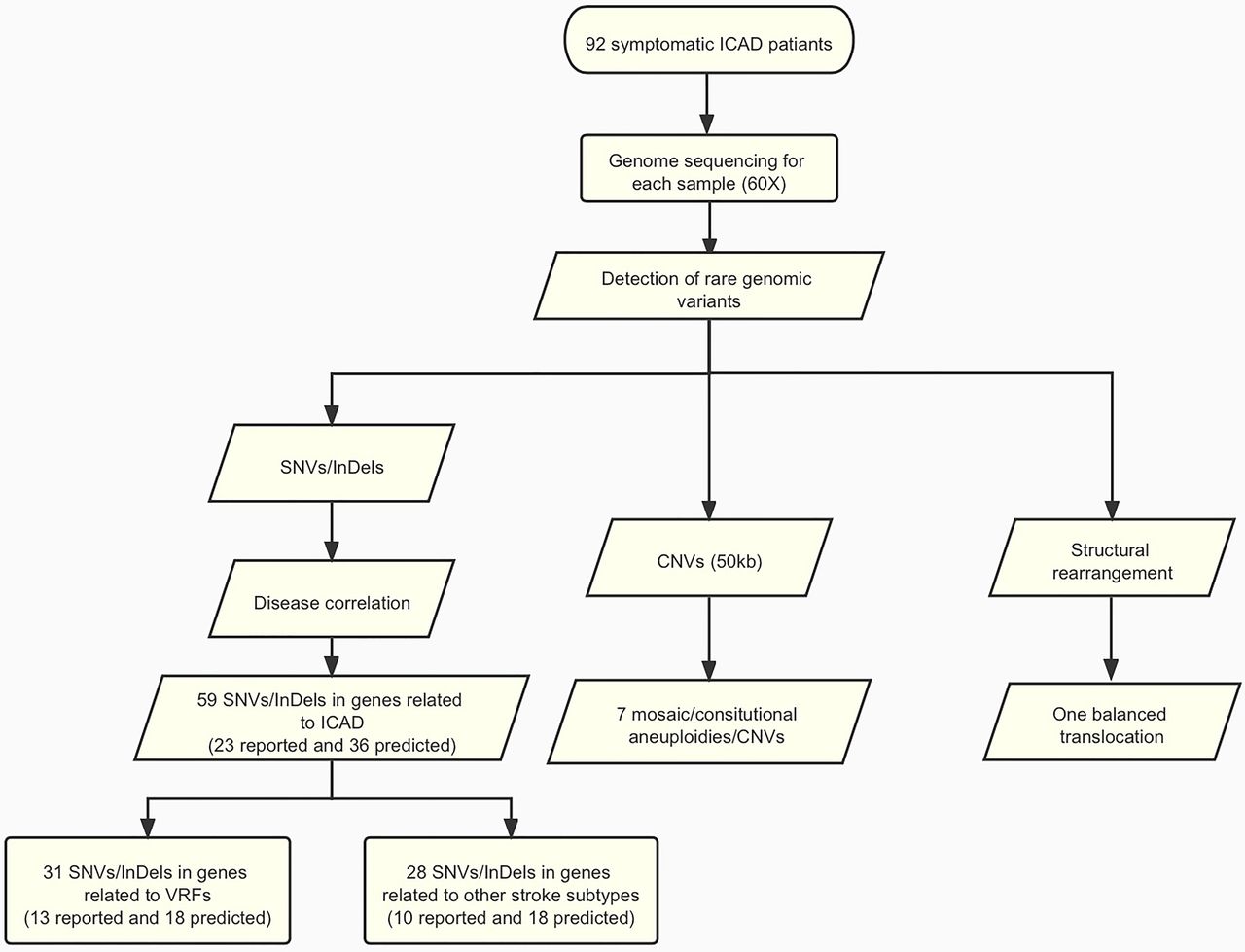
After detection of SNVs/InDels, we selected rare variants previously reported or predicted as potentially deleterious by in silico prediction software for further investigation. There were approximately 164 reported or predicted deleterious SNVs/InDels per case. After correlation to ICAD, we identified 59 reported or predicted to be deleterious SNVs/InDels in 48 patients, including 23 SNVs/InDels reported in the literature and 36 predicted to be deleterious (figure 1 and online supplemental eTable 3). None of them presented in the 126 local non-ICAD Chinese subjects. All 59 variants were successfully verified by Sanger sequencing (online supplemental eTable 2), providing a 100% of concordant rate of variants identified.

Flowchart of this study. Detailed methods and results are described in the main text. A total of 92 patients with symptomatic ICAD were submitted to genome sequencing (60-folds). Variant analysis was performed and identified 59 SNVs/InDels (23 reported and 36 predicted). In addition, genome-wide analysis revealed seven likely ICAD-related aneuploidies (loss of Y chromosome)/CNVs (>50 kb) and one balanced translocation. CNV, copy number variant; ICAD, intracranial atherosclerotic disease; InDel, small insertion/deletion; SNV, single-nucleotide variant; VRF, vascular risk factor.
Among 59 SNVs/InDels, 31 SNVs from 25 patients (25/92, 27.2%) were in genes related to conventional VRFs: (1) 5 SNVs were related to hypertension in four patients; (2) 15 SNVs were related to dyslipidaemia in 16 patients; and (3) 11 SNVs were related to diabetes in 11 patients. Thirteen of the 31 variants (41.9%) were reported previously. Of these 25 patients with ICAD, 18 (72%) had VRFs at baseline which are all or partly consistent with the VRF-related varaints (online supplemental eTable 4).
For the other 28 SNVs/InDels from 33 patients, they were in genes related to other stroke subtypes, involving NOTCH3, GLA, NF1, COL4A2, COL4A1 and RNF213 genes.19–21 Three variants in the NOTCH3 gene (R544C, V237M and L1518M) identified in this study were previously reported in patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) syndrome,22–24 and one heterozygous E66Q variant in GLA was reported in women with atypical Fabry disease(online supplemental eTable 3). However, none of these patients were diagnosed with CADASIL or Fabry disease at index stroke onset. In addition, we also detected 13 missense SNVs of RNF213 gene in 14 (15.2%) out of 92 patients. Five of these variants were previously reported in patients with moyamoya disease or ICAD. For instance, variants D4863N and E4950D in RNF213 (which were previously reported in Chinese patients with moyamoya disease and intracranial major artery stenosis/occlusion25) were identified in three patients with ICAD. However, none of these 14 patients was diagnosed with moyamoya disease by clinical or imaging profile.
We further retrieved the MAFs of each variant of the 59 SNVs/InDels from databases of NCVD, ChinaMAP and gnomAD, and all databases show extremely low minor allele frequencies in the Chinese population. In particularly, 20 out of 59 variants were not presented in ChinaMAP, a database with over 20 000 Chinese subject.
CNVs and chromosomal structural rearrangements potentially contributing to ICAD
We identified seven mosaic/consitutional aneuploidies/CNVs and one balanced translocation t(15;22) potentially related to ICAD in eight patients (figure 1 and online supplemental eTable 5). All these genomic variants were absent in our local subjects and the public database in general population (gnomAD). All mosaic/consitutional aneuploidies/CNVs were confirmed by CMA, and the balanced translocation was confirmed by both karyotyping and Sanger sequencing.
Among the 66 male patients, we identified mosaic aneuploidies (loss of Y chromosome (LOY)) in 5 cases with mosaic levels ranging from 12.4% to 50.0% (figure 2A). The age at stroke onset of these five subjects ranged from 64 to 75 years. Moreover, GS detected one constitutional and three mosaic deletions in two cases, of which two deletions in two subjects were found to be potentially associated with ICAD, including one 5 Mb constitutional heterozygous deletion in 10q11.22q11.23 (figure 2B) involving the GDF2 gene in one patient and one mosaic deletion of 2p23.3 involving the DNMT3A gene in another patient.

CNVs detected by GS. (A) Mosaic aneuploidies (loss of Y chromosome) detected by GS: mosaic level is around 50%. The X-axis indicates the genomic location of chromosome Y in human genome reference (GRCh37/hg19), while the Y-axis shows the copy number of chromosome Y. The mean copy ratio is shown by the red line. (B) A 5 Mb constitutional heterozygous deletion in 10q11.22q11.23 detected by GS (shown in the left side) and confirmed by CMA (right side). In the left side, the X-axis indicates the genomic location of the human genome reference (GRCh37/hg19), while the Y-axis shows the copy number. The heterozygous deletion is indicated by a red line with a pair of an arrow and the band region shown in the bottom. In the right side, probe distribution on the CMA platform with the candidate region reported by low-pass GS highlighted in red. CMA, chromosomal microarray analysis; CNV, copy number variant; GS, genome sequencing.
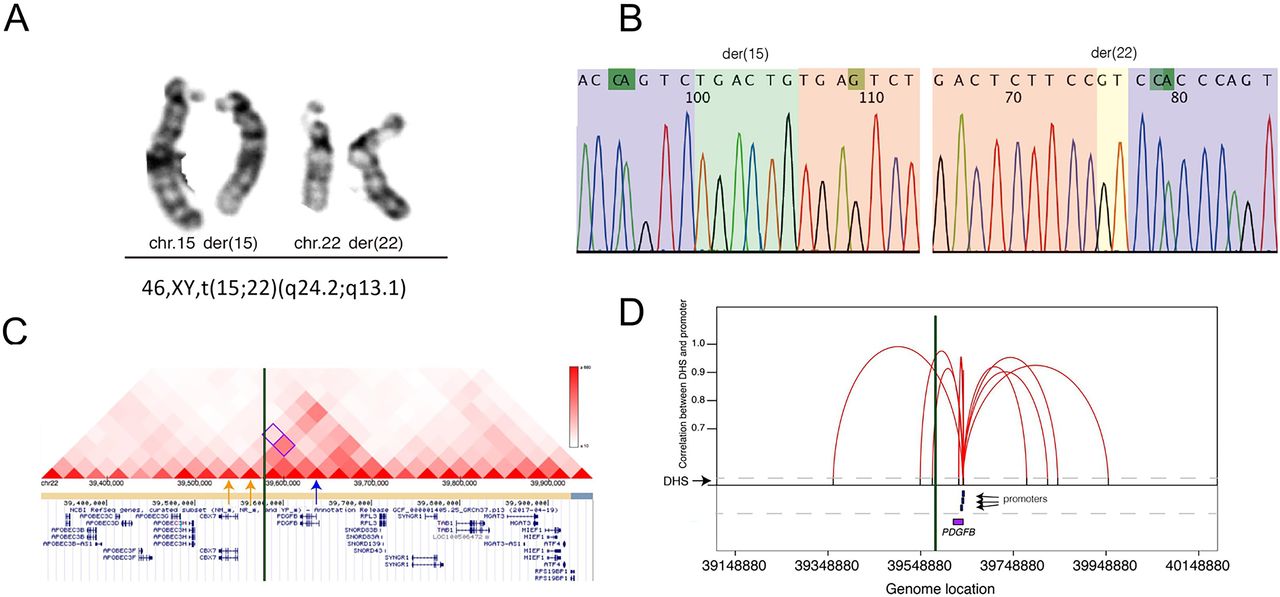
Furthermore, there were three insertions and one balanced translocation detected in four cases (online supplemental eTable 6), and only the translocation was likely related to ICAD. In a patient with 78% intracranial atherosclerotic stenosis, we found 46,XY,t(15;22)(q24.2;q13.1) involving chromosomes 15 and 22. Although neither breakpoints disrupted a known disease-causing gene, one breakpoint in chromosome 22 was located in a topologically associated domain involving the PDGFB gene, likely resulting in a disruption of the interaction between regulatory elements and gene (figure 3C,D).26 The protein encoded by the PDGFB gene plays an essential role in blood vessel development.27 In addition, disruption of the interactions between enhancers and promoters of gene PDGFB were also confirmed by the analysis of DNase I hypersensitive sites that are the hallmarks of regulatory DNA (figure 3C,D). Thus, this balanced translocation was suspected to result in ectopic expression of gene PDGFB during the development of ICAD.
{kind=link}
{kind=link}
{kind=link}
Identification of a balanced translocation in one patient. (A) Validation result by G-banded chromosome analysis (karyotyping) suggested a balanced translocation: 46,XY,t(15;22)(q24.2;q13.1). (B) Sequencing chromatograms of the breakpoint junctions. Sequences (from Sanger sequencing) of chromosomes 15 and 22 are highlighted in purple and pink, respectively. Inserted sequences and microhomozygous involved in the breakpoint junctions of the derivative chromosomes are also highlighted in green and yellow, respectively. (C) Breakpoints likely disrupted topological associated domain involving the PDGFB gene. Visualisation (http://www.kobic.kr/3div/) of interaction between gene PDGFB and the other locations in the reported database. Distribution of topological associated domains (triangles in different scales of red indicate different levels of interactions). The breakpoint junction is shown in the green vertical bar. The location of PDGFB gene is indicated by a blue arrow, while two windows potentially involved in the interaction of PDGFB gene are indicated by two yellow arrows. (D): disruption of the interactions between DHSs and promoters of PDGFB by the breakpoints. Figure indicates the diagram of the cross-cell-type correlation between distal DHSs and promoters of gene PDGFB based on the reported map. X axis represents the genomic coordinate of each element (such as gene and promoter), while Y axis shows the value of each correlation (r>0.9, reflected by a red line) between distal DHSs (indicated by the black bar) and promoters (blue bar) of gene PDGFB (purple box). The breakpoint junction is also shown in the vertical bar. DHSs, DNase I Hypersensitive sites.
Gene enrichment analysis
For gene enrichment analysis, 14 pathways/biological processes were found to be significantly enriched (online supplemental eTable 7). For instance, nine genes were significantly enriched in cholesterol transport (GO:0030301, multitest adjusted p value=10−10.56), while five genes were significantly enriched in acylglycerol homeostasis (GO:0055090, multitest adjusted p value=10−6.36).
Discussion
In this study, we investigated the potential genetic basis of symptomatic, high-grade ICAD by GS in 92 unrelated Chinese patients by identifying rare and potentially deleterious genomic variants in genes related to VRFs and/or other stroke subtypes. We identified 59 rare reported or predicted to be deleterious SNVs/InDels, 7 mosaic/constitutional aneuploidies/CNVs and 1 balanced translocation potentially contributing to ICAD in 54.3% (50/92) of the patients.
To investigate the genetic background of common diseases, common variants previously revealed by GWAS usually have very small effect sizes. Therefore, in contrast to the Common Disease–Common Variant Hypothesis, the Common Disease-Rare Variant Hypothesis might provide evidence.28 This hypothesis states that common polygenic diseases may be caused by the convergence of multiple, rare variants in the same gene or multiple genes. In the current study, we used GS to comprehensively investigate the role of rare genomic variants (SNVs/InDels, mosaic/constitutional aneuploidies/CNVs and chromosomal structural rearrangement) in a cohort of patients strictly phenotyped by clinical and imaging presentations.
The probably higher prevalence of conventional VRFs (hypertension, diabetes and dyslipidaemia) in Chinese patients who had a stroke/TIA, compared with Caucasian counterparts, may partly explain the higher prevalence of ICAD in Chinese. In addition to the possibly less stringent VRF management in Chinese than in Caucasians in more developed western countries,29 there may be a genetic basis as well. Therefore, our finding of rare and deleterious VRF-related variants in Chinese patients with ICAD indicated the role of such genetic basis overlapping with ICAD. In addition, we also identified rare SNVs/InDels in genes previously reported to cause SVS, which is known to share some risk factors (eg, the conventional VRFs) with ICAD, and both stroke types benefit from VRF management (eg, antihypertensive therapy).30 Furthermore, regarding the variants in the RNF213 gene, which was previously known in relation to moyamoya disease but recently discovered in patients with intracranial arterial stenosis/occlusion,31 our study confirmed that the well-known R4810K variant was absent in all patients. However, we identified 13 rare variants in the RNF213 gene in 14 patients. None of these patients was diagnosed of moyamoya disease, based on detailed clinical and imaging workups; thus, it indicates ICAD might also share the genetic basis with moyamoya disease. Overall, our finding of rare and potentially deleterious genomic variants in genes reported in VRFs and other stroke subtypes also supported the shared genetic aetiology, which needs verification in future studies.
There may be doubts regarding the lack of ‘controls’ in the GS analysis, for example, ‘healthy’ family members for segregation analysis or other controls without stroke/ICAD for disease correlation study. However, ICAD is an evolving disease that the possibility for future ICAD development in healthy subjects could not be ruled out, and that early-stage ICAD (eg, intimal thickening) in apparently healthy subjects may not be picked up in imaging exams, both of which may cause confounding in the analyses with controls. Therefore, sex-matched and age-matched healthy subjects might not be suitable controls for comparison. In this study, we retrieved the MAF of each variant in large-scale databases of both at-risk subjects and general populations. First of all, none of the detected variants presented in the 126 local Chinese subjects without ICAD who underwent GS. In addition, 20 variants were not present in the Chinese population (ChinaMAP, sample size of over 20 000). It indicates that applying sex-matched and age-matched healthy controls with similar sample size for an association study would not be a proper method for investigating the contribution of rare genomic variants to ICAD.
Finally, another important new finding of this study was the detection of CNVs and chromosomal structural rearrangements by GS, which has not yet been well studied in ICAD. Among the genomic variants identified, seven mosaic/constitutional aneuploidies/CNVs and one structural rearrangement (8.7%, 8/92) potentially contributed to the development of ICAD. For instance, among the cases with mosaic LOY, experimental reduction of UTY gene (in Y chromosome) expression had been associated with perturbation of pathways causing atherosclerosis,32 and LOY in blood cells had been associated with increased risk of cardiovascular diseases and mortality in ageing men.32 Therefore, mosaic loss of chromosome Y is a possible genetic contributor to ICAD. In addition, two deletions were found to be associated with ICAD as for the involvement of GDF2 and DNMT3A genes, respectively. Disruption of GDF2 might cause pulmonary hypertension and atherosclerosis.33 In a mice model, loss-of-function mutations of DNMT3A could lead to accelerated atherosclerosis and convergent macrophage phenotype,34 and individuals with somatic mutations of the DNMT3A gene in haematopoietic cells had been reported with an increased risk of cardiovascular diseases in a human study.35 Furthermore, the balanced translocation 46,XY,t(15;22)(q24.2;q13.1) was likely to affect the expression of PDGFB gene (figure 3C), which is known to be associated with impaired recruitment of pericytes to blood vessels in endothelial cells, and mice carrying hypomorphic Pdgfb alleles might develop brain calcifications with age-related expansion.27 36 Overall, none of the CNVs and structural rearrangement was reported in general populations in public databases. Thus, our study provides evidence that rare CNVs and structural rearrangements could play a role in ICAD development, but further functional validation is warranted.
Further gene enrichment analysis revealed that the candidate ICAD-related genes were significantly enriched in the pathways or biological processes involved in lipoprotein metabolism and cellular lipid catabolic process. These pathways and different lipids/lipoproteins are involved in atherogenesis. For instance, retention of apoB-containing lipoproteins (low density lipoprotein (LDL), intermediate density lipoprotein (IDL) and very low-density lipoprotein (VLDL)) in the arterial intima plays an important role in initiating atherosclerosis.37 Subsequently, lipoproteins undergo modifications and ultimately trigger a series of maladaptive responses that accelerate further lipoprotein retention and lead to plaque progression.37 Therefore, variants in genes involved in these pathways/processes might be an underlying genetic mechanism of ICAD.
Limitations
Overall, this study revealed ICAD-related genomic variants including SNVs/InDels, mosaic/constitutional aneuploidies/CNVs and structural rearrangements. However, there were still limitations. First, the diagnosis of symptomatic ICAD in this cross-sectional study only represented a snapshot of this evolving disease, while the ICAD lesion might have presented long before the index stroke onset and the severity of luminal stenosis in ICAD would change over time or with treatment. This also explained why we did not correlate the onset age and the severity of luminal stenosis in ICAD with the variants identified. On the other hand, we could not rule out the possibility that some individuals would develop ICAD and perhaps subsequent ischaemic stroke in years. However, this limitation is probably inevitable in investigating the genetic background of an evolving disease like ICAD. In addition, with extended candidate gene selection criteria (involvement of genetic variants possibly contributing to VRFs and/or other stroke subtypes) and relatively loose criteria for defining functional variants, the current study might have overestimated the rate of patients with ICAD with genomic variant. One the other hand, we acknowledged that our analysis had excluded the potential for identifying new candidate genes associated with ICAD by limiting the SNVs/InDels of interest to genes that were previously associated with VRFs and/or other stroke subtypes. Lastly, further studies with large-scale sample size and/or functional analysis are needed to verify the current findings and the proportion of genetic factors in leading to the predisposition to ICAD, as ICAD is a multifactorial disease attributed to inheritable, environmental and pathophysiological factors. Moreover, it would be ideal to include patients with ICAD from European populations, as well as Chinese/European subjects with carotid atherosclerosis, for comparison to further illustrate the role of genetic factors underlying ICAD in different populations.
Conclusions
Our GS study identified 59 rare SNVs/InDels, 7 mosaic/constitutional aneuploidies/CNVs and 1 balanced translocation potentially contributing to ICAD in 54.3% (50/92) of symptomatic patients with ICAD. Overall, our study demonstrated the potential role of rare genomic variants in the development of ICAD in Chinese patients.
Data availability statement
Data are available upon reasonable request. Anonymised data can be available for qualified investigators upon request to the corresponding authors.
Ethics statements
Patient consent for publication
Ethics approval
The study protocol was approved by the ethics committee of the Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee (reference number 2014.582-T).
Acknowledgments
We appreciate all participants in this study.
References
Supplementary material
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
MS and XL contributed equally.
Contributors TWL and ZD had the full access to all of the data in the study and took responsibility for the integrity of the data and the accuracy of the data analysis. MS and XL contributed equally to this study in data analysis and manuscript written-up. TWL, ZD and KWC designed the study and reviewed, edited and approved the final version. YL, ZC, YC, TC, BYMI, VHLI, YOYS, FSYF, SHM, KM, AYYC, LWCA, HL, AYL and VCTM collected the data.
Funding This study is funded by Kwok Tak Seng Centre for Stroke Research and Intervention, the National Natural Science Foundation of China (31801042), the Health and Medical Research Fund (04152666 and 07180576), 2018 Shenzhen Virtue University Park Laboratory Support Special Fund (YFJGJS1.0) for Key Laboratory for Regenerative Medicine, Ministry of Education (Shenzhen Base) and The Chinese University of Hong Kong Direct Grant (2019.051 and 2019.033).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.