Article Text

Abstract
Background Tenecteplase (TNK) possesses several pharmacological characteristics superior to conventional alteplase (rt-PA), with well-established safety and efficacy profile in Caucasians. There exists controversy over the optimal dose of intravenous rt-PA for East Asians with acute ischaemic stroke (AIS). Current study aimed to determine the safety dose range of recombinant human TNK tissue-type plasminogen activator (rhTNK-tPA) for patients with AIS in China.
Methods This multicentre, prospective, randomised, open-label, blinded end-point, phase II study compared three tiers of 0.1, 0.25, 0.32 mg/kg rhTNK-tPA (to a maximum of 40 mg) with standard 0.9 mg/kg rt-PA (to a maximum of 90 mg) in patients who were eligible for intravenous thrombolysis. The safety outcome were symptomatic intracranial haemorrhage (sICH) within 36 hours.
Results Between May 2018 and February 2020, 240 patients were randomly assigned to four group, 4 of whom did not receive study treatment. The intention-to-treat analysis included 236 patients. There was no difference in the improvement on National Institutes of Health Stroke Scale at day 14 in the 3 tiers and control group (63.3%, 77.2%, 66.7% vs 62.7%). The number of sICH was 3 of 60 (5.0%) in the 0.1 mg/kg group, none in the 0.25 mg/kg group, 2 of 60 (3.3%) in the 0.32 mg/kg group and 1 (1.7%) of 59 in the rt-PA group. There were no significant between-group differences in severe adverse events.
Conclusions Similar to the Caucasians, rhTNK-tPA was well tolerated in Chinese patients with AIS at all doses administered within 3 hours of symptom onset. The dose-efficacy profile of rhTNK-tPA needs to be established with future investigations.
Trial registration number NCT04676659.
- stroke
- thrombolysis
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
INTRODUCTION
Intravenous thrombolysis with alteplase (rt-PA) is recommended as the standard treatment for eligible patients with acute ischaemic stroke (AIS) in guidelines.1 2 The short half-life of rt-PA limits its use, especially in patients who need endovascular thrombectomy. Tenecteplase (TNK) is a genetically modified form of human tissue plasminogen activator (tPA) that has advantages over rt-PA on three aspects: (1) higher fibrin specificity that make it possible to reduce haemorrhagic complications, (2) greater resistance to plasminogen activator inhibitor-1 that may possess higher efficacy in clot lysis and (3) longer serum half-life that allows for single-bolus administration.3 4
TNK has been used for treatment of Caucasians patients with AIS at doses of 0.1, 0.25, 0.4 or 0.5 mg/kg.5–8 Different doses were chosen in two phase III studies. The EXTEND-IA TNK study came to the conclusion that TNK usage before thrombectomy was associated with a higher rate of reperfusion and better functional outcome than rt-PA in patients with large vessel occlusion (LVO) AIS treated within 4.5 hours after symptom onset.9 TNK (0.4 mg/kg) did not confer better outcome over the 0.25 mg/kg dose.10 The Norwegian tenecteplase stroke trial (NOR-TEST) showed a similar safety profile between the TNK (0.4 mg/kg) and rt-PA in patients with mild AIS.11 12 There is a lack of consensus on the safe and effective dose of TNK in patients with AIS.
Moreover, the optimal dosage of rt-PA for East Asians is still in debate.13–16 As a potential competitor of rt-PA, it is necessary to determine the dose range of optimal safety in Chinese patients with AIS.
This study, Tenecteplase Reperfusion therapy in Acute ischemic Cerebrovascular Events (TRACE), compared three different doses of recombinant human TNK tissue-type plasminogen activator (rhTNK-tPA) with standard rt-PA treatment in patients with AIS within 3 hours from symptom onset. Patients were chosen randomly to receive either rhTNK-tPA or rt-PA. rhTNK-tPA, approved for treating acute myocardial infarction in China, had the same terminal amino acid sequence and different production process to the TNK made by Boehringer (Metalyse) and Genentech (TNKase) which was used in previous studies (online supplemental tables 1 and 2).
Supplemental material
Methods
Study design
The TRACE was a multicentre, prospective, randomised, open-label, blinded end-point, phase II study done in 22 stroke units in China (online supplemental table 3) between May 2018 and February 2020. The protocol was approved by the local institutional review board and all patients or legal representatives had given informed consent.
Patients
Patients were eligible if they were 18 years of age or older, were diagnosed of AIS with measurable deficits on the National Institutes of Health Stroke Scale (NIHSS) 4–25 (both included), haemorrhage ruled out by non-contrast computer tomography (NCCT) scan, were admitted within 3 hours of symptom onset which refers to ‘last known to be well’, living independently (prestroke modified Rankin Scale (mRS)≤2 or without history of stroke) and had no contraindications to intravenous thrombolysis (online supplemental table 4).
Randomisation and masking
Patients were randomly assigned (1:1:1:1) to rhTNK-tPA 0.1, 0.25, 0.32 mg/kg (to a maximum of 40 mg) or rt-PA 0.9 mg/kg (to a maximum of 90 mg) using a strict central block randomisation system (over the internet). A total of 4 lots of rhTNK-tPA (Guangzhou Recomgen Biotech) and 25 lots of rt-PA (Boehringer Ingelheim) were provided free of charge to patients and used within the validity period. The treatment was conducted openly. Outcomes were assessed by an independent investigators who were blinded during all procedures and well trained with NIHSS and mRS evaluation.
Procedures
Eligibility for intravenous thrombolysis was assessed at the emergency department and a baseline NIHSS examination was performed. The planned number of patients to be enrolled in each dosage tier was 60. rhTNK-tPA (Guangzhou Recomgen Biotech, China; 1.0×107 IU/16 mg per vial) was given as a single, intravenous bolus (over 5–10 s) immediately after randomisation. Maximum dose was 40 mg. 10% dose of rt-PA (Actilyse; Boehringer Ingelheim, Germany; 50 mg, 20 mg per vial) of the active control group was given as bolus and the remainder over 1 hour (maximum dose was 90 mg). Other treatments were carried out adhering to established clinical principles and medical practice guidelines. Bridging thrombolysis plus endovascular thrombectomy was allowed but was excluded from per protocol analysis.
Outcomes
Safety outcomes were incidences of symptomatic intracranial haemorrhage (sICH) within 36 hours (defined as European Cooperative Acute Stroke Study, ECASS III),17 asymptomatic intracranial haemorrhage identified by routine repeat brain imaging within 90 days, systematic bleeding at 90 days, deaths from any cause and adverse events/serious adverse events (SAEs) within 90 days. The primary efficacy outcome was defined as the proportion of subjects with improvement on NIHSS of ≥4 points or a score ≤1 at day 14.18 19 The secondary efficacy outcomes included (1) the proportion of subjects with mRS ≤1 point at 90 days; (2) the proportion of subjects with mRS ≤2 point at 90 days and (3) ordinal distribution of mRS at 90 days. An independent data monitoring committee (iDMC) maintained surveillance of patient safety. An iDMC meeting was held to decide whether to discontinue the study if three cases of sICH occurred in any group. The clinical events committee which was not involved in the execution of the study verified and adjudicated all clinical endpoints based on clinical symptoms, laboratory tests and imaging data.
Statistical analysis
All patients randomised to receive study treatment will be included in the final analysis for safety and clinical outcome (intention-to-treat analysis (ITT)). The per-protocol (PP) population was defined as all patients who received any dose of study drug and met all the inclusion and exclusion criteria. The last observation carried forward was used for patients who withdrew from the study and counted as lack of data. Proportions were used for categorical variables, and means with SD or median with IQRs were used for continuous variables. Differences in efficacy outcomes among four groups were analysed. The proportion of NIHSS ≥4 points or a score ≤1 at day14 were compared by logistic regression. OR and the 95% CI were used as significance measures. Ordinal distribution of mRS at 3 months was analysed. All tests were two-sided, and a p value of 0.05 or less was considered statistically significant. All statistical analyses were performed using SAS V.9.4 software.
Results
Baseline characteristics
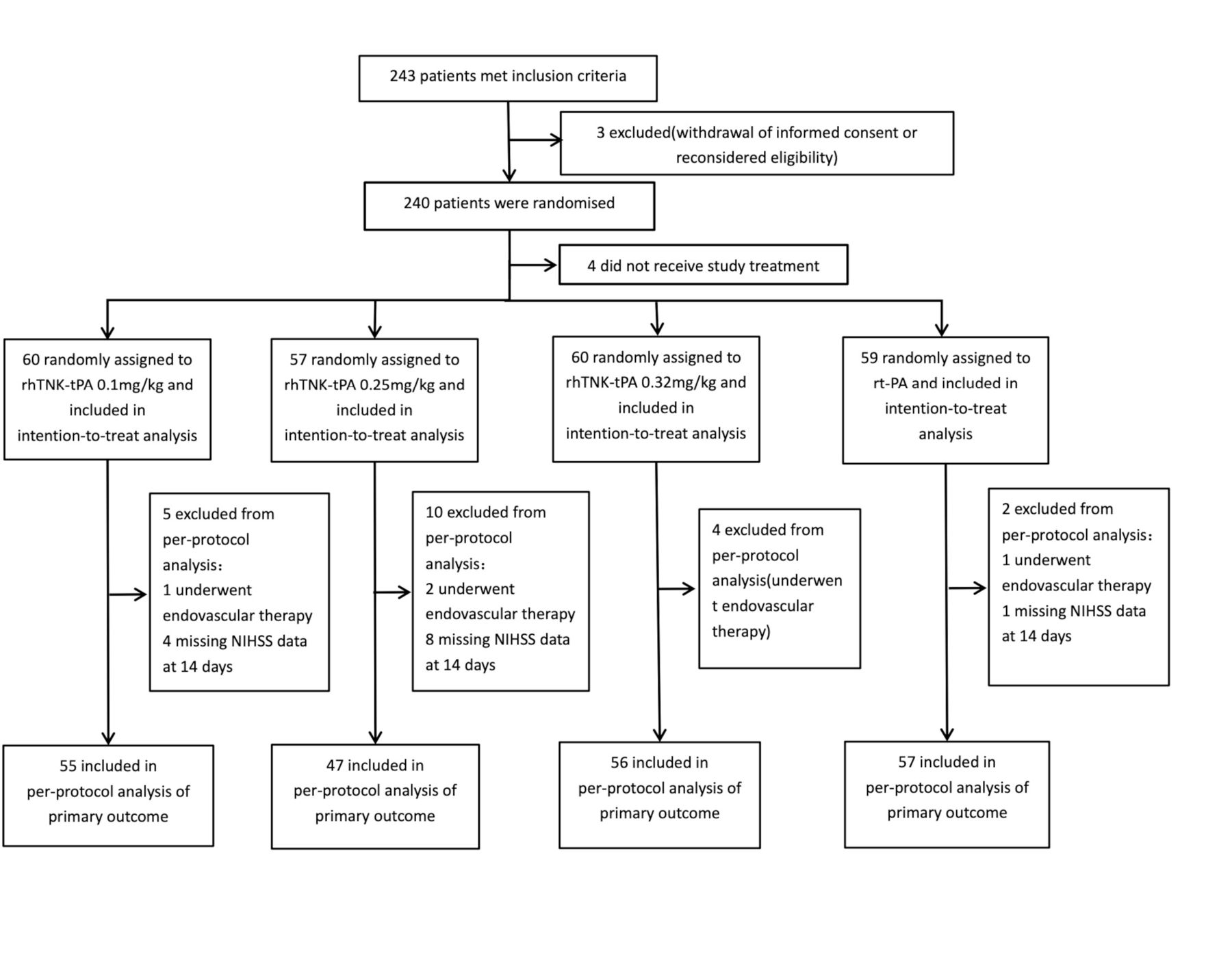
Between May 2018 and February 2020, 243 patients with AIS were enrolled at 22 clinical sites. Three patients were excluded because informed consents were withdrawn after enrolment. Out of 240 patients randomised, 4 did not receive study treatment. Sixty patients were assigned to receive 0.1 mg/kg of rhTNK-tPA, 57 were assigned to receive 0.25 mg/kg of rhTNK-tPA, 60 were assigned to receive 0.32 mg/kg of rhTNK-tPA and 59 to receive 0.9 mg/kg of rt-PA. Patients (21) missing NIHSS data at 14 days or undergoing endovascular therapy were excluded from the PP population (figure 1). The four groups were well balanced regarding baseline characteristics (table 1). The mean ages were 62.4 (11.1), 64.3 (12.8) and 64.8 (12.1) for rhTNK-tPA groups vs 66.5 (12.6) of the rt-PA group and the median NIHSS were 7.0 (5.0–10.0), 8.0 (5.0–12.0) and 7.5 (6.0–12.0) for rh-TNK-tPA group vs 8.0 (5.0–12.0) of the rt-PA group.
Characteristics of patients at baseline
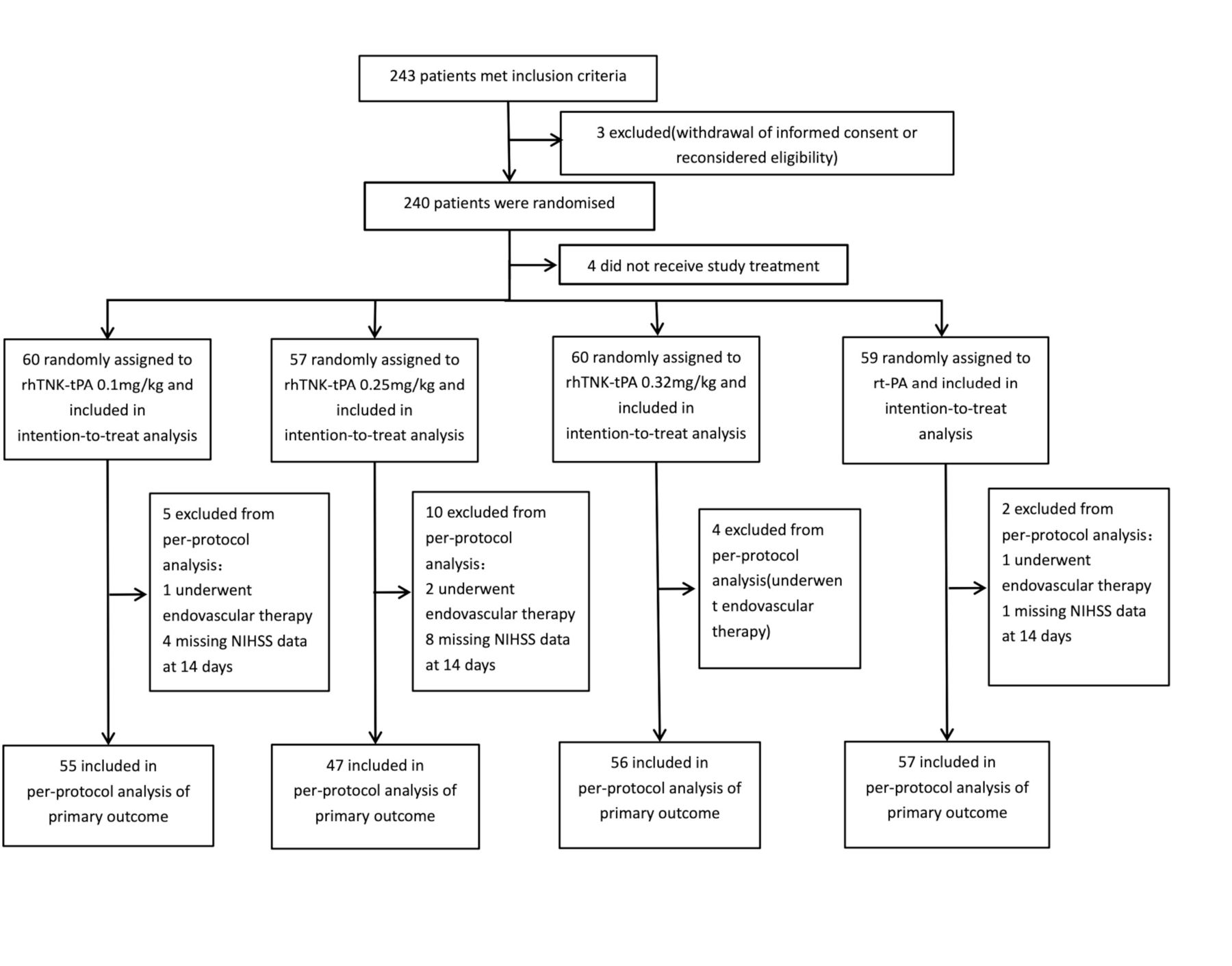
Flowchart.
Safety outcomes
Table 2 summarised the haemorrhagic complications, death and SAE encountered in the trial. No datum was missing in safety outcomes. The sICH events were observed in 3 (5%), 0, 2 (3.3%) and 1 (1.7%) patients in the three-tier rhTNK-tPA groups and the rt-PA group, respectively. The incidence of fatality with 0.1 mg/kg of rhTNK-tPA was 6 (10%), 0.25 mg/kg was 1 (1.8%), 0.32 mg/kg was 5 (8.3%) and 0.9 mg/kg of rt-PA was 6 (10.2%). SAEs (life-threatening, resulting in prolonged or recurrent hospitalisation, disability, dysfunction, lead to abnormalities) were observed in 12 (20%), 7 (12.3%), 11 (18.3%) and 14 (23.7%) patients in the 0.1 mg/kg, 0.25 mg/kg 0.32 mg/kg of rhTNK-tPA group and the rt-PA group, respectively. SAEs classified by system organ were listed in online supplemental table 7. No significant difference was found in all safety outcomes.
Prespecified safety outcomes and other serious adverse events
Efficacy outcomes
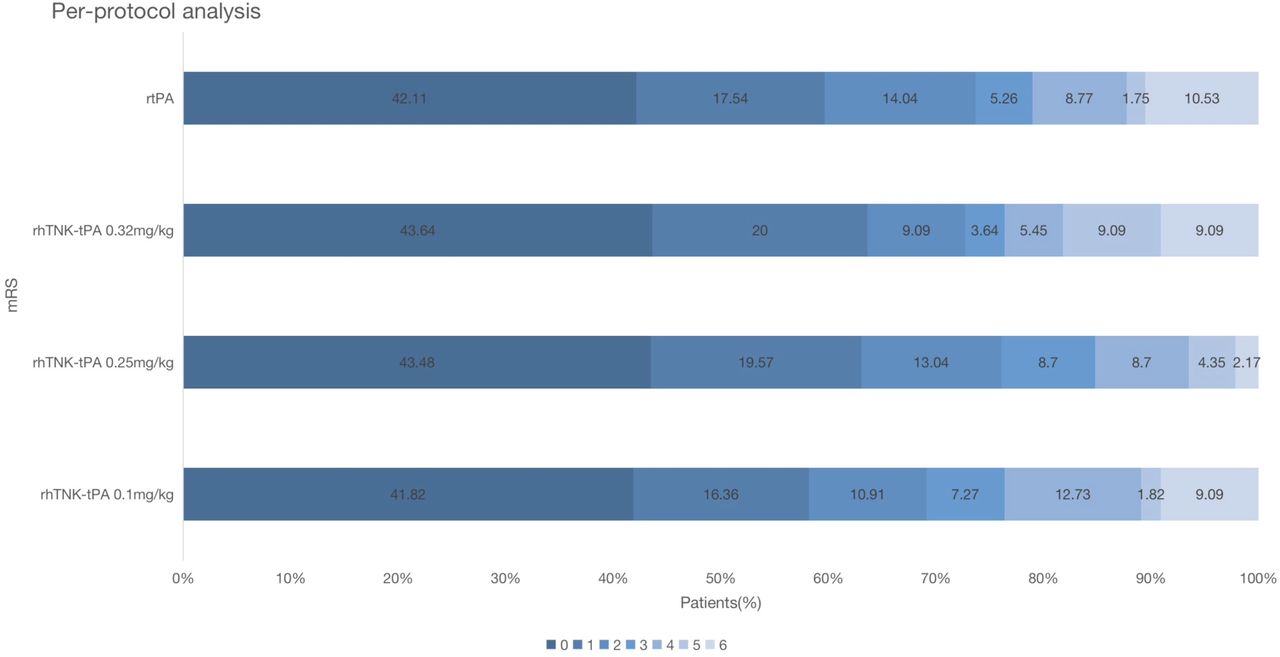
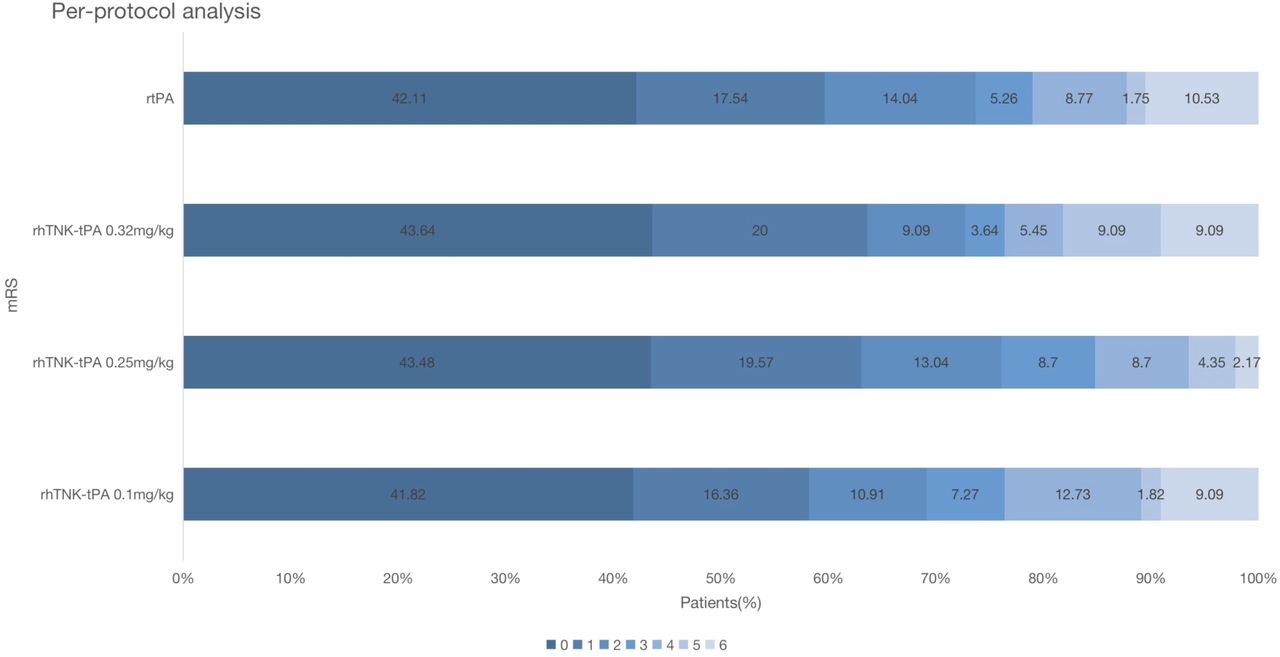
Table 3 displayed the efficacy outcomes of NIHSS at 14 days and mRS at 3 months. Data of improvement on NIHSS at 14 days were missing in 20 patients: 11 patients for death within 14 days and 9 patients for telephone follow-up (online supplemental table 5). In the ITT analysis, the proportion of primary efficacy outcome in the 0.1 mg/kg, 0.25 mg/kg and 0.32 mg/kg of rhTNK-tPA group and the rt-PA group was 63.3%, 77.2%, 66.7% and 62.7%, respectively. The mRS distribution is shown in figure 2. No significant difference of any efficacy outcomes was observed between the three-tier rhTNK-tPA groups and the rt-PA group (online supplemental table 6). The data from PP population were consistent with the ITT population.
Primary efficacy outcome and secondary efficacy outcome in the intention-to-treat and per-protocol populations at 90 days
{kind=link}
{kind=link}
mRS at 90 days in the per-protocol population. mRS, modified Rankin Scale.
Discussion
TNK was indicated non-inferiority to rt-PA based on meta-analysis.20 21 On the other hand, the optimal dose has varied between 0.25 mg/kg and 0.40 mg/kg. rhTNK-tPA used in TRACE study has the same terminal amino acid sequence and different production process to TNK used in previous studies. In this study, patients with AIS treated with 3-tiers of rhTNK-tPA showed similar rates of improvements on neurological deficits and sICH, compared with patients treated with rt-PA. The dose tier of 0.25 mg/kg indicated more proportion of improvement on neurological deficit, lower proportion of death and no sICH event.
A pilot dose-escalation study for the safety of TNK in AIS provided the initial experience with dose tiers of TNK and discarded 0.5 mg/kg.5 The 0.4 mg/kg tier was considered unsafe in a prematurely terminated randomised clinical trial as 3 (16%) of 19 patients suffered sICH.6 However, the NOR-TEST showed relatively low rates (9%, 47 of 549) of sICH in patients receiving 0·4 mg/kg of TNK.11 The EXTENT-IA TNK part 2 used the dose of 0.4 mg/kg in patients with large-vessel occlusion and the rates of sICH was 4.7% (7 of 150).10 In TRACE study, 3.3% patients suffered sICH in the 0.32 mg/kg dose tier, and no sICH in the 0.25 mg/kg dose tier was the same as in the rt-PA group (1.7%). The low rates of sICH in our study might be related to the baseline characteristics of patients and the improved quality of medical practice. The mean age was younger and the median NIHSS was lower in our study than most previous studies. The study population of NOR-TEST was the closest to the population of our study, which used 0.4 mg/kg of TNK in patients with AIS within 4.5 hours. The mean age of TNK group in NOR-TEST was 70.8 (14.4), and the median NIHSS score was 4 (2-7). No new safety concern of rhTNK-tPA was observed in this study.
The intrinsic characteristics of the drug recipient and extrinsic characteristics associated with environment and culture that could affect the results of clinical studies carried out in regions. The low-dose (0.6 mg/kg) rt-PA strategy to treat patients with AIS in East Asian countries derived from the traditional concept that East Asians had a higher prevalence of sICH than Caucasians patients.22 Despite the Enhanced Control of Hypertension and Thrombolysis Stroke Study (ENCHANTED) did not support low-dose rt-PA to standard-dose rt-PA in predominantly Asian patients (63.2%)13 or in Chinese population,23 small sample data of East Asian countries, as represented by China, Japan and South Korea, did not coincide with each other.14–16 A recently pool analysis of two observational registries demonstrated that low-dose rt-PA was associated with significant reduction of sICH and non-inferior performance in efficacy for moderate stroke patients in China.24 As a target-modified variant of rt-PA, TNK would face the same problem. The data from local population may be required to determine whether TNK used in East Asians is as effective and safe as in Caucasians. The results of current study of rhTNK-tPA in China were comparable to the study of TNK in Caucasians. The 0.25 mg/kg dose tier performed better on improvement of neurological deficits (77.2% vs 66.7% and 62.7%) and excellent functional outcomes (mRS score ≤1, 63.6% vs 62.1% and 59.3%) than other groups, which shall be selected as the study dose in future trials of greater sample sizes. In our study, 3 hours within stroke attack was chosen because rt-PA was not yet approved to use in the 3–4.5 hours window after stroke attack, when our study was conceptualised. During the recruitment stage of our study, the results of the phase III trial of rt-PA used in 3–4.5 hours after AIS were reported and rt-PA received the label for use in 4.5 hours within stroke attack.25 A prospective TRACE II study will investigate the efficacy and safety of rhTNK-tPA for AIS within 4.5 hours.
TNK is a promising agent for AIS treatment.26 27 TRACE study gave us the confidence to carry out further investigations of rhTNK-tPA. Two phase III trials (TRACE II and TRACE III) of rhTNK-tPA for injection versus standard medical treatment for AIS within 4.5 hours and for AIS due to LVO with perfusion mismatch up to 24 hours of symptom onset will be launched in the coming year. Other ongoing trials of TNK whose results are highly anticipated include NOR-TEST2 (NCT03854500), TASTE (ACTRN12613000243718), ATTEST2 (NCT02814409), TWIST (NCT03181360) and AcT (NCT03889249).
Limitations
This study had several limitations, such as small sample size, an open-label design and a maximum dose lower than 0.4 mg/kg. Due to the limitation of sample size, the evidence collected from this study was still inadequate for an unambiguous recommendation of optimal dose. Although the maximum dose of rhTNK-tPA is 0.32 mg/kg in our study, the efficacy and safety of rhTNK-tPA seem to be either comparable if not superior to published studies. In addition, NCCT is effective before intravenous thrombolysis for patients with AIS within 4.5 hours of onset according to the guidelines. We did not use advanced imaging to identify LVO at baseline. The upcoming TRACE III trial will assess the effect of rh-TNK for patients with AIS with LVO in the late time window (4.5–24 hours after stroke onset).
Conclusion
Our results showed that intravenous rhTNK-tPA, given within 3 hours of symptom onset, is a well-tolerated option in patients with AIS in China. A dose of 0.25 mg/kg may be suggested for future efficacy studies in Caucasians patients with AIS based on the results of contemporary clinical studies. The efficacy dose of rhTNK-tPA in East Asians needs to be investigated with further investigation.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and the principles of the Declaration of Helsinki.
Acknowledgments
We would like to thank all the patients and investigators who participated in the study.
References
Supplementary material
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @yilong
Contributors YoW, XZ and YiW contributed to study design. SL administered the study. ZW, ZL, HC, DW, YS, WD and HZ contributed to data collection. YP did the data analysis. SL wrote the first draft. All authors read and approved the final manuscript.
Funding This study is supported by the National Natural Science Foundation of China (81870905), the National Key R&D Program of China (2017YFC1308204), the National Science and Technology Major Project (2017ZX09304018) and the Beijing Municipal Administration of Hospitals Incubating Program (PX2018022).This study was sponsored and funded by Guangzhou Recomgen Biotech Co., Ltd.
Disclaimer Guangzhou Recomgen Biotech Co., Ltd. had no role in the design and conduct of the study; collection, management, analysis and interpretation of data, preparation, review or approval of the manuscript and decision to submit the manuscript for publication.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.