Article Text

Abstract
Background Mendelian stroke causes nearly 7% of ischaemic strokes and is also an important aetiology of cryptogenic stroke. Identifying the genetic abnormalities in Mendelian strokes is important as it would facilitate therapeutic management and genetic counselling. Next-generation sequencing makes large-scale sequencing and genetic testing possible.
Methods A systematic literature search was conducted to identify causal genes of Mendelian strokes, which were used to construct a hybridization-based gene capture panel. Genetic variants for target genes were detected using Illumina HiSeq X10 and the Novaseq platform. The sensitivity and specificity were evaluated by comparing the results with Sanger sequencing.
Results 53 suspected patients of Mendelian strokes were analysed using the panel of 181 causal genes. According to the American College of Medical Genetics and Genomics standard, 16 likely pathogenic/variants of uncertain significance genetic variants were identified. Diagnostic testing was conducted by comparing the consistency between the results of panel and Sanger sequencing. Both the sensitivity and specificity were 100% for the panel.
Conclusion This panel provides an economical, time-saving and labour-saving method to detect causal mutations of Mendelian strokes.
- stroke
- technology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Mendelian strokes are a group of monogenic disorders caused by rare non-synonymous variants often leading to small vessel disease and intracerebral haemorrhage.1 It causes nearly 7% of strokes and is also an important aetiology of cryptogenic stroke.2 The prevalence of Mendelian stroke is always underestimated for the following reasons: varying phenotypic expressions, in the absence of the characteristic manifestations, variable disease penetrance and lack of knowledge about the diseases by doctors.3 4 Recently, based on the data of Exome Aggregation Consortium database (ExAC database), it is estimated that the prevalence of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) can reach 3.4/1000, which is 100-fold higher than what would be expected based on the current CADASIL prevalence estimations of 2–5/100 000.5–8 Identifying the genetic abnormalities in Mendelian stroke is important as it would facilitate clinical diagnoses, therapeutic management and genetic counselling. The main advantage of next-generation sequencing (NGS) is the inexpensive production of a large amount of sequencing data.9 In order to study the influence of genetic factors on stroke and assist in the diagnosis of Mendelian stroke, we designed a customised panel using NGS technology to detect genetic variants related to cerebrovascular disease, such as causal mutations for Mendelian stroke, risk factor-related genes mutations, genetic variants associated with disease susceptibility and drug metabolism. This panel also included hundreds of other candidate genes in stroke-related pathways. Therefore, application of this panel could increase the ability of Mendelian stroke diagnoses in a cost-efficient manner and deepen the understanding on mechanism of stroke.
Objectives
Create a gene panel for testing of Mendelian strokes.
Evaluate the analytical validity of the gene panel.
Methods
Searching of causal genes for Mendelian stroke (genes list of panel)
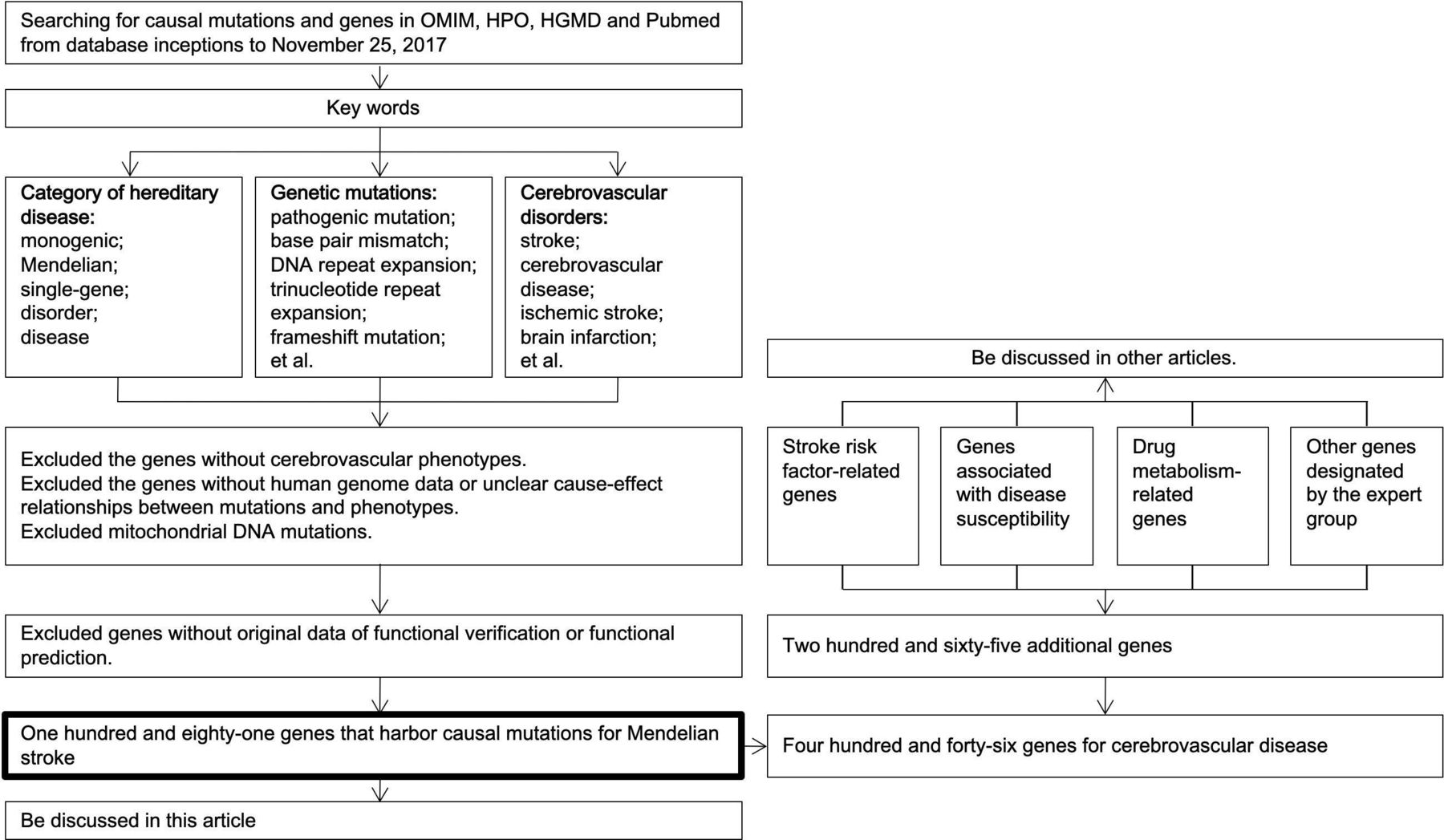
In order to construct the panel, a comprehensive search for causal mutation and genes of Mendelian strokes was performed on Online Mendelian Inheritance in Man, Human Phenotype Ontology, Human Gene Mutation Database professional databases and the PubMed in November 2017. The search was conducted using keywords of the following three items and were combined by the Boolean logical operator AND: category of hereditary disease (including “monogenic”, “Mendelian”, “single-gene”, “disorder”, and “disease”); genetic mutations (including “pathogenic mutation”, “base pair mismatch”, “DNA repeat expansion”, “trinucleotide repeat expansion”, “frameshift mutation”, “gain of function mutation”, “gene amplification”, “gene duplication”, “genomic instability”, “microsatellite instability”, “germ-line mutation”, “in/del mutation”, “loss of function mutation”, “mutagenesis, insertional”, “mutation, missense”, “point mutation”, “sequence deletion”, “gene deletion”, “sequence inversion”, “suppression, genetic”, and “synthetic lethal mutations”); and cerebrovascular disorders (including “stroke”, “cerebrovascular disease”, “ischemic stroke”, “brain infarction”, “transient ischemic attack”, “TIA”, “intracerebral hemorrhage”, “subarachnoid hemorrhage”, “aneurysm”, “moyamoya disease”, “moyamoya syndrome”, “artery dissection”, “arterial-venous malformation”, and “systematic embolic”) (figure 1).
{kind=link}
Screening genes of the panel. HGMD, Human Gene Mutation Database; HPO, Human Phenotype Ontology; OMIM, Online Mendelian Inheritance in Man.
Three authors carried out the above manual search, and the inclusion criteria for the search results were items or literatures that (1) reported a causal mutation for cerebrovascular disease, (2) described the detailed phenotype of the patients, (3) contained functional verification or prediction for the mutations. Items or literatures that were conducted using non-human materials or did not report clear cause–effect relationships between mutations and phenotypes were excluded. Any disagreement on inclusion of the items or literatures was reviewed by a senior neurologist and resolved by consensus from senior neurologists.
Afterwards, two authors independently extracted the genes and mutations in the qualified search items (category of hereditary disease; genetic mutations; cerebrovascular disorders) and literatures using a standardised form (online supplementary table s1). Furthermore, a senior expert reviewed the two lists of genes and mutations, and resolved the disagreements based on the agreement of inclusion criteria.
Supplemental material
The panel also contained genes and genetic variants that were associated with stroke risk factors or susceptibility, as well as some drug metabolism-related genetic variants of stroke therapy, and other genes designated by the expert group (figure 1). These genes were added for scientific research purposes and will be discussed in other studies.
Construction of the panel
According to the provided list of genes, a SureSelect Target-Enrichment panel was designed using the online tool SureDesign (https://earray.chem.agilent.com/suredesign, Agilent technologies, Santa Clara, California, USA). The panel mainly covered coding exons of the genes, and also covered some genetic variants that were associated with ability of drug metabolism. The panel was designed under default parameter settings of SureDesign.
DNA preparation and NGS
For each participant, DNA was isolated from peripheral leukocytes using DNA Isolation Kit (Bioteke, AU1802, Beijing, CHN). DNA libraries were prepared using KAPA Library Preparation Kit (Kapa Biosystems, KR0453, Wilmington, Massachusetts, USA) following the manufacturer’s instructions. Target fragments were captured using the designed panel. Paired-end reads (150 bp) were generated by HiSeq X10 or Novaseq (Illumina, San Diego, California, USA).
Bioinformatics analysis
Trimmomatic (V.0.36) was applied to remove adapters and low-quality reads.10 Afterward, qualified reads were aligned to the human reference genome sequence from the University of California, Santa Crus Genome Browser Database (UCSC) (hg19, downloaded from http://genome.ucsc.edu/) using the Burrows-Wheeler Alignment tool.11 Genetic variants were called using the Genome Analysis Tool Kit, V.4.0.12.0 joint calling function under best practice guidance.12–14 A hard filter (depth ≥9, genotype quality score ≥15) was applied for quality control of the variants. Genetic variants with allele frequency <1% in 1000 genome, Genome Aggregation Database (gnomAD) and Exome Sequencing Project V. 6500 (esp6500) were further annotated by Clinical Interpretation of Genetic Variants by the 2015 American College of Medical Genetics and Genomics (ACMG)-Association for Molecular Pathology (AMP) Guidelines (InterVar) and dbscSNV under the guidelines of the ACMG and the AMP.15 16
Variant classification
Candidate variants were estimated for pathogenicity based on the ACMG guidelines.17 The special criteria are as follows: (1) whether the variant was reported by functional or family segregation study, previously; (2) the type of the variant (eg, nonsense mutation, frameshift mutation or splicing mutations); (3) variant frequency in the ExAC, gnomAD and 1000 Genomes Project databases; (4) conservation of the altered residue and (5) family segregation studies and de novo mutation. According to this information, a variant was further categorised into one of the following categories: pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign or benign.
After the patient was submitted for examination, the variants were interpreted for pathogenicity according to the database retrieval at that time. However, due to the continuous updating of the database and the frequency data of different ethnic groups, the latest database was introduced to interpret these variants again. All of the interpretation processes are in strict accordance with ACMG guidelines.17
Informed consent and clinical diagnoses
In order to explore the effectiveness of the panel, patients who were highly suspected to be affected by Mendelian strokes were recruited. Informed consent was provided by patients from April 2018 to February 2019. Detailed clinical features, cerebrovascular risk factors and family history were collected by neurologists. The diagnostic criteria for Mendelian stroke phenotype-based algorithm which had referred to a literature and made some revisions were2: (1) patients suffered from a stroke or transient ischaemic attack with unknown etiopathogenic causes; (2) the presence <3 conventional vascular risk factors (such as hypertension, hypercholesterolaemia, diabetes mellitus, hyperhomocysteinaemia, obesity, hyperuricaemia, atrial fibrillation and smoking), young age at onset (≤55 years), positive familial history or specific clinical features of Mendelian stroke (such as angiokeratoma, O'Sullivan sign, et al); (3) an age of onset >55 years old, positive familial history, highly suspected Mendelian stroke by experienced neurologists. Patients that conform to (1) and any two of (2) or any two of (3) were highly suspected to be affected by Mendelian stroke and were enrolled in this study.
Confirmation
Sanger sequencing (ABI 3730 DNA Analyzer, Thermo Fisher Scientific, Massachusetts, USA) was performed to verify the genetic variants of pathogenic/likely pathogenic/VUS. The primers were designed using Primer Premier V.5.0 (Premier Biosoft, USA) and PCR was performed to amplify the fragments covering the mutated sites on a LifeECO Thermal Cycler TC-96/G/H (b)C(Bioer Technology, CHN). The PCR products were further purified using agarose gel electrophoresis and then sequenced. Sanger sequencing results were analysed by Chromas Lite V.2.01 (Technelysium, Tewantin, QLD, Australia).
Diagnostic testing
Diagnostic testing was performed to evaluate the specificity and sensitivity of the panel by investigating the consistency between panel and Sanger sequencing. The reference sequences were retrieved from the hg19 human genome in the UCSC genome browser, and Sanger sequencing results were aligned and compared with reference sequences using Lasergene SeqMan Pro software (Version 7.1.0, DNASTAR, Madison, USA). For each of the 78 point mutations (online supplementary table s2), the diagnostic testing was performed by comparing the base calling between panel and Sanger sequencing in the fragment of 41 bp that ranges from 20 bp upstream to 20 bp downstream of the point mutation. For each of the 3 In/Dels (online supplementary table s2), the diagnostic testing was performed by comparing the base calling between panel and Sanger sequencing in the upstream or downstream 40 bp fragments of the In/Del mutation.
Supplemental material
Results
Construction of the panel
A total of 181 genes that were reported to harbour causal mutations for Mendelian strokes were identified after a thorough search on the online databases in November 2017 (online supplementary Table S3). Additionally, genes and genetic variants that were associated with stroke risk factors or susceptibility, as well as some drug metabolism-related genes of stroke therapy, and other designated genes by the expert group were also included (table 1). Consequently, the total targeted region size of the panel was 1.93 Mbp, and it covered coding exons of 446 genes (online supplementary Table S3).
Supplemental material
Composition of the gene panel
High-quality NGS data of the panel
The performance of the panel was evaluated by conducting NGS on patients that were highly suspected to be affected by Mendelian strokes (table 2).
Overview on the sequencing data
The accuracy of the panel was investigated by comparing the results of panel and those of Sanger sequencing. In the 181 genes that harbour causal mutations for Mendelian strokes, a total of 81 pathogenic, likely pathogenic, and VUS variants were suspected to be candidate causal mutation of the patients after the first round of variants interpretation which was performed immediately after genetic testing (online supplementary Table S2). In the first round of variants interpretation, we refer to the databases from April 2018 to February 2019, of which 52 out of 53 patients’ data were analysed in 2018, and one in February 2019. All of these mutations were verified by Sanger sequencing.
To evaluate the sensitivity and specificity of the panel, the consistency between base calling of panel and Sanger sequencing for a 40 bp or 41 bp fragment around each of the aforementioned 81 variants was explored. Among these fragments, only one additional common SNP (rs199685642) with a homozygous alternative-allele genotype was found, which is 19 bp downstream of the PKD1c.1138C>T mutation (Figure S1). Therefore, the diagnostic test was conducted using a total of 82 genetic variants. The results of panel and Sanger sequencing were consistent that both the sensitivity (82/82) and specificity (3239/3239) were 100%.
Supplemental material
Updates in the second round of variants interpretation
We performed variants interpretation again based on the latest database in March 2020. Compared with the first round, it was found that the pathogenic, like pathogenic and VUS variants dropped from 15 to 0, from 8 to 4 and from 58 to 12, respectively, although all of these two rounds of analyses were both conducted in strict accordance with the guidelines of ACMG (online supplementary Table S2 and table 3). These four likely pathogenic variants in the second round of analysis are consistent with the clinical phenotype of autosomal dominant inheritance, so Mendelian stroke can be diagnosed clinically.
Summary of the genetic test results of 53 probands analysed in March 2020
Discussion
Stroke is one of the leading causes of mortality and disability, annually affecting 10.3 million people worldwide.18 Stroke is usually regarded as a multifactorial and polygenic disease,19–26 yet hereditary factors could induce stroke by monogenic, polygenetic or epigenetic modes.27 Mendelian stroke is a rare but important cause of stroke. Although it is rare, diagnosis is very important for individual patients. It can carry out predictive test for other family members and prenatal examination. Up to now, there is no report about using a gene panel by NGS platform to detect Mendelian stroke.
In this study, we not only designed a comprehensive hybridization-based target-enrichment gene panel but also established a corresponding NGS and bioinformatics analysis pipeline that could be applied to find causal genetic mutations of Mendelian stroke. The pipeline exhibited excellent performance with high-quality data (average depth ≥194×, 20×coverage ≥96.5%). In comparison with Sanger sequencing, both the sensitivity and specificity of the panel was 100%, and the lowest coverage of the mutation that was confirmed by Sanger sequencing was 9×, suggesting the high accuracy and credibility of the panel.
This panel with phenotype-based algorithm provided an effective method to find genetic abnormalities in Mendelian strokes for which likely pathogenic mutations were identified in 4 out of 53 participants based on the pathogenicity defined by the second round of interpretation of variants conducted in the latest database. Combined with clinical phenotype analysis, 4 out of 53 patients were diagnosed with Mendelian stroke, around 7%, similar to the previous study conducted using Sanger sequencing,2 which suggests that Mendelian stroke should be paid attention to in patients with clinical suspected Mendelian stroke since these genetic disorders may provide insights to study the underlying biological mechanisms of a complex disease like stroke. Particularly, the application of this panel could save time, labour and costs for diagnoses and differential diagnoses mainly because it allows simultaneous genetic testing for a total of 446 genes. Conducting Sanger-sequencing-based genetic testing for such a large number of genes is expensive and difficult, given the clinical complexity and heterogeneity of Mendelian strokes. Conversely, in comparison with whole-genome or whole-exome sequencing, both the difficulties of bioinformatics analysis and associated expense was reduced for the panel, because only genes that have been reported to harbour causal mutations were included.
Due to the continuous updating and supplement of the population genomic data for the interpretation of genetic variants, the classification of the mutations was different between the two rounds of variant interpretation. Sixty-five variants that were rated VUS and above in the first round were rated to be likely benign or benign in the second round. The decrease of pathogenicity rating of these variants was mainly caused by population genomics data accumulation and the allele frequency of these variants were found to be higher than previously known. Therefore, when explaining the report to the clinician, it is proposed that the analysis of the pathogenic variants should be strictly compared with the clinical situation before the clinical pathogenic diagnosis. Special attention should be paid for the continuous update of the database and the subsequent change in the pathogenicity rating and judgements, and clinical diagnosis based on genetic testing should be more cautious for patient.
In this work, we applied Illumina HiSeq X10 and the Novaseq platforms in high-throughput DNA sequencing because of their high accuracy and the well-controlled expense . In spite of the advantages, the disadvantage is the high duplication rate in the sequencing data due to the PCR amplification during library construction. Processing these duplicated reads consumes computation power and might prolong the time length of bioinformatics data analysis. However, in this work, the computation power is adequate to handle the sequencing data of the panel because we focus on a limited region of the genome and processing the duplicates would not waste too much time.
There are also a few limitations for this panel. First, although careful literature searching and review was conducted, the panel only included a majority but not all of the causal genes for Mendelian strokes. In the recent years, new causal genes of Mendelian stroke have been emerging, such as COLGALT1.28 And the panel requires updating to include new causal genes in the future. Second, while aiming to identify the causal mutations of Mendelian strokes, this study mainly focused on the CDS of targeted genes. Therefore, the effect of intronic and intergenic mutations may not be fully explored. Third, in order to simultaneously screen 181 genes of Mendelian strokes for clinical diagnosis and 265 genes for research purpose related to stroke, we captured more than four hundred genes in parallel. Although the overall detection effect is satisfactory, it was inevitable that the sequencing depth or coverage of certain individual gene was not high enough. Fourth, the small sample size might potentially introduce sampling bias. A multicentre study with a larger sample size will reduce the bias, and help to evaluate the efficiency of the panel more fully.
In summary, we presented a panel for genetic testing of Mendelian strokes. For pathogenic/likely pathogenic/VUS genetic variants, the results of panel and Sanger sequencing were consistent. This panel provided an effective tool to support diagnoses and genetic counselling for Mendelian strokes. In addition, this panel can also be used for scientific research related to stroke.
Acknowledgments
We gratefully thank Xiaoning Chen (Running Gene) for her assistance with data collection, and Jun Sun (Running Gene) for her help with the panel design. Both have provided the corresponding author with permission to be named in the manuscript. And we thank all the doctors for supplying patients in the department of neurology, Beijing Tiantan Hospital.
References
Footnotes
FF and ZX are joint first authors.
WL and YW contributed equally.
Contributors YW and WL had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: YW, WL and HL. Searching of Causal Genes: YS and SC. Diagnostic testing and statistical analysis: FF and ZX. Bioinformatics analysis: ZX. Drafting of the manuscript: FF, ZX, YS, HW, WL, HL and YW. Critical revision of the manuscript for important intellectual content: HL. Study supervision and organization of the project: WL and YW.
Funding The Ministry of Science and Technology of the People’s Republic of China (2016YFC0901001, 2016YFC0901002, 2016YFC0901004, 2017YFC1310901, 2017YFC1310902, 2018YFC1311700, 2018YFC1311706) National Science and Technology Major Project (2017ZX09304018) Beijing Municipal Commission of Health and Family Planning (No.2016-1-2041, SML20150502). Beijing Municipal Science & Technology Commission (D171100003017002).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval This study was approved by the local ethical committees of Beijing Tiantan Hospital, Capital Medical University.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement No data are available. This paper is a research protocol, which is mainly used to explain the principle and availability of detection technology, so no data are provided.