Article Text

Abstract
The discovery of targeted drugs heavily relies on three-dimensional (3D) structures of target proteins. When the 3D structure of a protein target is unknown, it is very difficult to design its corresponding targeted drugs. Although the 3D structures of some proteins (the so-called undruggable targets) are known, their targeted drugs are still absent. As increasing crystal/cryogenic electron microscopy structures are deposited in Protein Data Bank, it is much more possible to discover the targeted drugs. Moreover, it is also highly probable to turn previous undruggable targets into druggable ones when we identify their hidden allosteric sites. In this review, we focus on the currently available advanced methods for the discovery of novel compounds targeting proteins without 3D structure and how to turn undruggable targets into druggable ones.
- big data
- artificial intelligence
- novel drugs
- 3D structure
- undruggable targets
- hidden allosteric sites
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
As we all know, there are shortcomings in the traditional drug research and development field: (1) research and development cycle is too long: a new drug can take 14–15 years or more to develop1; (2) research and development cost is expensive (US$2.6 billion per new drug); (3) the success rate of drug research and development is very low. Although conventional methods such as combinatorial chemistry (CC) and high-throughput screening (HTS) can be used for effective drug development, they are cumbersome processes and require huge investment. In the past decade, as traditional approaches to drug development have become increasingly difficult, computer-aided drug design (CADD) had been used to improve the success rate of drug discovery. And it has become a powerful tool for the development of small molecule therapies with higher hit rates than HTS and CC.2 When both HTS and CADD methods were used to screen a novel inhibitor of protein tyrosine phosphatase-1B, a key target for type II diabetes, CADD showed higher hit rates.3
CADD is a method based on computer chemistry to predict and calculate the relationship between ligand and receptor through computer simulation, to carry out the design and optimisation of lead compounds. After years of efforts and exploration, the CADD method has gradually become mature and played an important role in drug design and development. These include proteomics, genomics, structural biology, bioinformatics, chemoinformatics and HTS.4 CADD usually has two different calculation methods for drug design: ligand-based drug design (ie, indirect drug design, LBDD) and structure-based drug design (ie, direct drug design, SBDD).5 LBDD is generally preferred only when compounds with activity data are available; SBDD relies on information about the real three-dimensional (3D) structure of proteins or 3D models reconstructed by homologous modeling.6 This article mainly introduces SBDD.
Protein Data Bank (PDB) is one of the most commonly used repositories of biomolecular structure information, most of which is determined by X-ray crystallography, while the smaller group is determined by cryogenic electron microscopy (Cryo-EM) and nuclear magnetic resonance techniques (NMR) spectrum7; this provides support for SBDD. However, it is difficult to obtain the actual structure of many specific proteins which cannot be expressed or purified at a certain scale. When the target protein does not have a well-resolved 3D structure, the homologous modelling method can be used to predict its structure and guide further experimental work, which is also a relatively reliable method.8 9 When the hidden allosteric sites located on proteins with 3D structure are identified, it is possible to turn previously undruggable targets into druggable ones. Based on our previous research experience in artificial intelligence (AI)-assisted drug discovery, we review the above methods and successful cases to provide a theoretical basis for subsequent research work.
Target protein database
The structure and function of protein are an important research content in life science. The 3D folding structure of protein natural folding determines its function. And it is very important for drug research and development. At present, the methods to obtain the 3D structure model of real protein include: X-ray crystal diffraction techniques, NMR and Cryo-EM techniques. With rapid development of these technologies, more and more protein structures are being analysed. So, researchers established many protein 3D structure information databases.
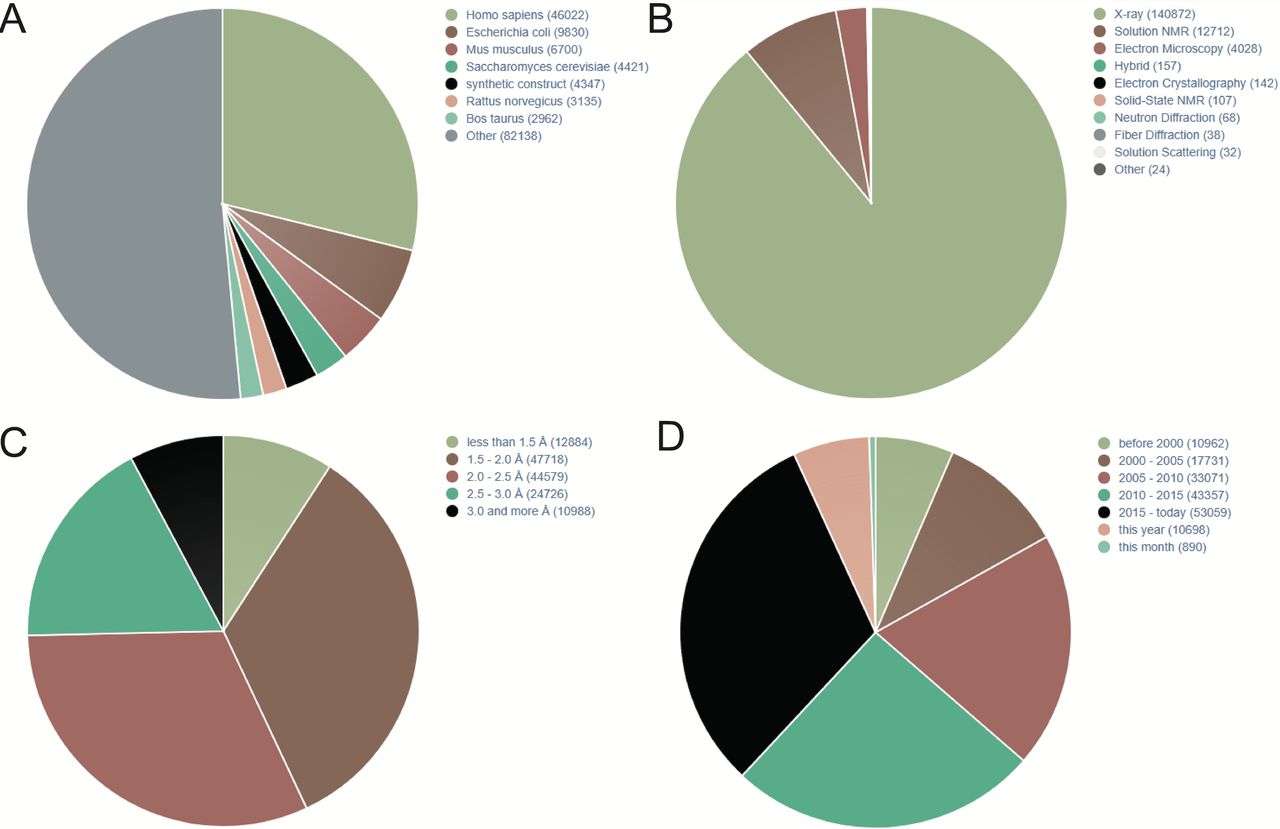
PDB is a commonly used protein database, which was founded in 1971. It is a single global repository, which stores the 3D structures of biological macromolecules and their complexes determined by experiments. It is also the first open-access digital resource in the biological field.10 In addition to the 3D coordinates, the PDB file also contains the general information needed for all sedimentary structures and the information unique to the structure determination method.11 Figure 1 shows that according to PDB statistics, 3D protein structures are rapidly increasing. In addition, according to different research purposes, the researchers further constructed a more detailed database of protein receptors, as shown in the following table.(table 1)12

Statistics of three-dimensional (3D) protein structures in Protein Data Bank (PDB; December,2019). (A) 3D protein structures distributed in different species. (B) 3D protein structures parsed by different methods. (C) The resolution of 3D protein structures in PDB. (D) Increasing trend of published 3D protein structures during last two decades. NMR, nuclear magnetic resonance.
Protein structure databases
Prediction of protein 3D structures
In recent years, with the development of genomics/transcriptomics/proteomics, more and more protein sequences have been identified. For proteins with no available 3D structure, the above method cannot be used for drug design. Therefore, researchers have designed a series of new methods to predict and construct protein 3D structure model based on bioinformatics. There are three common methods: (1) homologous modelling: the 3D structure of the target sequence is deduced by looking for its homologous protein. The commonly used software is SWISS-MODEL, which requires that a known structure with a consistency of the target sequence ≥30% as the template. For example, Dhanavade et al predicted the structure of cysteine protease, an important target of Alzheimer’s disease, by homologous modelling.13 (2) Folding recognition: also known as threading method, it is used to find proteins that have no significant homologous relationship with the target sequence but have the same structural folding type, and finally establish the structural model. The commonly used software is I-TASSER. It can start with amino acid sequences and construct a 3D structure model by recombining the fragments cut from the thread template, and then match the structure model with the known proteins in the functional database to predict the biological function of the target proteins.14 Spinocerebellar ataxia type 2 and 3 are two common autosomal dominant ataxia syndromes. Polyglutamine (poly-Q) is considered to be an important factor in the pathogenesis of the disease. Wen et al used I-TASSER to predict the structure of poly-Q.15 (3) Ab initio prediction: the 3D structure of the protein is predicted from the sequence itself. One can use AlphaFold,16 C-I-TASSER17 and so on. Both are a mixture of deep learning and traditional algorithms. Because of the large amount of calculation, this method is not commonly used.
In practice, we conducted many protein models by homologous modelling with I-TASSER. Based on these models, we have successfully screened agonists/antagonists for given proteins without any published 3D structure (unpublished data provided by Jingwei Jiang). Figure 2 shows the recently published crystal structure of extracellular domain of VISTA (PDB code: 6oil) and a corresponding VISTA homologous reconstructed model we constructed 2 years ago. We had obtained a very precise virtual VISTA 3D model (root mean SD=3.6 Å, structurally aligned to 6oil) for virtual screening of drug discovery. As a result, several agonists/antagonists lead compounds effective at nanomolar concentration have been discovered (unpublished data provided by Jingwei Jiang).
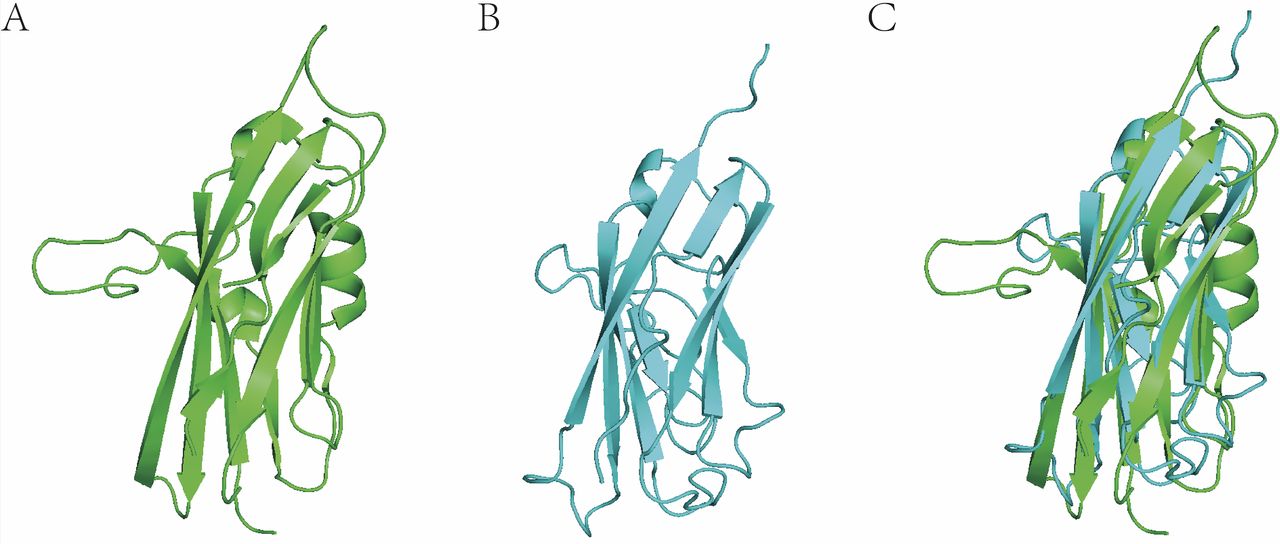
Three-dimensional (3D) protein structural alignment of VISTA (crystal structure) and homologous reconstructed model. (A) VISTA crystal structure (6oil, resolution=1.85 Å). (B) VISTA model. (C) 3D structural alignment between real 3D structure and 3D model (root mean SD=3.6 Å).
Homologous modelling is the most successful method in protein structure prediction at present. The boundary between folding recognition and homologous is becoming more and more blurred. Homologous modelling plays an important role in finding templates, while folding recognition uses the details of sequences to improve the accuracy of sequence alignment. Ab initio prediction of protein structure is an immature research field, but the development potential is very great, because this method is to predict the high-level structure of protein from its primary structure. In practical applications, homologous modelling is generally considered first. If the consistency between the template sequence and the target sequence is less than 30%, then the folding recognition method is recommended. If the result of structural evaluation is still not ideal, or the target sequence is less than 100 amino acids, ab initio prediction can be used. However, its performance is not good if the sequence is longer than 100 amino acids.
Prediction of ligand-binding sites on protein 3D models
The biochemical function of protein plays an important role in organism. However, the physical properties and physiological functions of many proteins have not been discovered. Proteins perform biological functions mainly by binding to their ligands. Identification of binding sites in proteins is essential for understanding their interactions with ligands as well as other proteins, and is very helpful for the discovery and rational design of therapeutic compounds.18 Although many methods have been developed to screen ligand-binding sites based on highly similar protein structures, those included in PDB are very limited. In fact, many proteins do not have a resolved crystal structure, using predicted low-resolution protein models to find and verify ligand binding sites has become a challenging problem. At present, with the continuous development of computer technology, we can make full use of the methods and advantages of AI to predict the binding sites of ligands on protein 3D models.
For some given amino acid sequences of benchmark proteins, the protein structure can be predicted by a variety of servers such as I-TASSER19 and PredictProtein.20 Subsequently, a variety of developed programs can be used to predict protein–ligand binding sites. For example, BSP-SLIM,21 as a method of ligand–protein blind docking using low-resolution protein structures, can use a given sequence to predict protein structure models through I-TASSER. The assumed ligand-binding sites were transferred from the holographic template structure of the model and the ligand–protein docking conformation was constructed by using the shape and chemical matching of the ligand and the negative image of the binding pocket. In addition, according to the Structural Classification of Proteins database, some similar or very distant homologous proteins can have common binding sites.22 According to this, FINDFITE23 can identify the ligand-bound structural template by algorithm and superimpose it on the structural model of distant or near homologous proteins to identify the hypothetical binding sites. On the other hand, the improved version of FINDSITEfilt of FINDSITE can elevate the accuracy and accuracy by filtering out the false-positive ligands in the thread recognition template.24 In this way, the ligand-binding sites on the 3D model of proteins without crystal structure can be predicted through a variety of available servers.
In practice, we have successfully applied large-scale structural alignment to predict a lot of ligand-binding sites and discover corresponding novel agonists/antagonists (unpublished data provided by Jingwei Jiang). Figure 3 shows the 3D structure of gasdermin D (GSDMD). We structurally aligned the human GSDMD (6n9o) to mouse GSDMD (6n9n) and predicted the polymerisation protein-binding pocket located on human GSDMD. Based on the predicted pocket, we have successfully discovered several novel antagonists inhibiting the polymerisation of human GSDMD both in vitro and in vivo within 1 month (unpublished data provided by Jingwei Jiang).
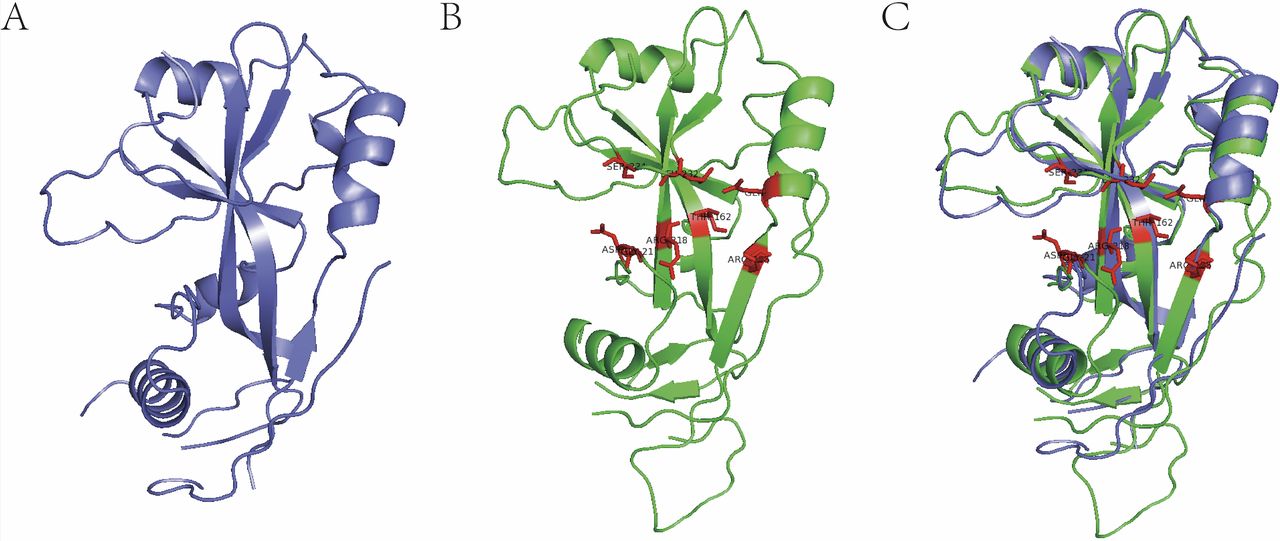
Identification of polymerisation pocket on gasdermin D (GSDMD) by 3D protein structural alignment. (A) Human GSDMD (6n9o). (B) Mouse GSDMD (6n9n), red amino acid residues are verified polymerisation-associated residues. (C) 3D alignment for the identification of corresponding polymerisation pocket residues on human GSDMD.
Undruggable targets and overcoming undruggable targets
The term ‘Undruggable’ is used to describe a protein that is not pharmacologically capable of being targeted; however, efforts are being made to turn these proteins into ‘druggable’, so it is more appropriate to describe it as ‘difficult to administer’ or ‘currently unavailable’.25 Over the past few decades, many pathways of tumourigenesis have been discovered with the efforts of scientists. Many proteins involved in cancer development, particularly kinases, provide drug targets. However, many proteins, such as RAS, MYC and p53, are considered as ‘Undruggable targets’. RAS mutations are early events in tumour progression, and there is substantial experimental evidence that sustained expression of RAS mutations is necessary for tumour maintenance. Despite more than 30 years of hard work, effective pharmacological inhibitors of RAS oncoprotein have not been approved by FDA (Food and Drug Administration) yet.26 MYC is a transcription factor that is involved in a variety of cancer-promoting programs and is often overexpressed in advanced stages of cancer, and various attempts have been made to overcome its lack of medicinally binding pockets.25 p53 is the most frequently altered gene in human cancer, with p53 mutations present in about 50% of all invasive tumours. It is clear that the p53 mutant protein is an important target, but it has traditionally been considered undruggable.27 These targets are due to their large or flat protein–protein interaction interfaces or lack deep protein-binding pockets.28 Therefore, overcoming these ‘so-called undruggable targets’ is one of the main challenges of drug discovery.
In recent years, researchers have proposed many new methods to solve this problem: (1) induce target protein degradation29; (2) block pathways downstream of the target30; (3) discover hidden allosteric sites, and so on. The discovery of hidden allosteric sites in these three approaches will greatly expand the types of existing drug targets and provide opportunities for the discovery of allosteric drugs. Therefore, the intervention of hidden allosteric sites is an effective method.
Hidden allosteric sites
In the design of targeted drugs, the combination of drugs and biological macromolecules is like the relationship between key and keyhole. An effective drug is like a key to the configuration of targeted macromolecules, so the question becomes to find the ‘keyhole’ and quickly match the ‘key’. Hidden allosteric sites are more challenging than allosteric sites because these sites are not visible in proteins even in the well-resolved crystal structure. Since the biological macromolecular protein in solution is not static, but dynamic, there is a combination of multiple conformations coexisting with different energies. The hidden allosteric site is a binding pocket that does not exist in the protein crystal structure, but it becomes available as the protein fluctuates. Such sites may have unknown biological functions and can be used as important targets for drug design.31
There are several methods to identify hidden allosteric sites32: (1) large-scale unbiased molecular dynamics (MD) simulations: in theory, a variety of protein conformations can be obtained by large-scale unbiased MD simulation of proteins. Several low-filled conformations obtained by MD simulation cannot be captured by experimental methods. These small conformations may have hidden allosteric sites, which can be used in the design of allosteric regulators. For instance, the discovery of allosteric sites for the b2-adrenalin receptors33; (2) combined ensemble-based docking and MD simulations, for example, andrographolide derivatives can inhibit the conformational transformation of RAS34; (3) accelerated MD simulations: compared with traditional MD simulation, accelerated MD by introducing a non-negative press or potential into the potential energy surface can promote the conformational transition between low energy states and effectively enhance the conformational sampling space of proteins, especially for proteins with slow time scale dynamics. Yang used this method to identify potential allosteric sites of interleukin-1 receptor35; (4) MD-based Markov state analysis: the Markov state model is based on a large number of MD simulations. The advantage of these models is that they can identify hidden allosteric sites that are usually located in small conformations, which can be captured from multiple simulations rather than a single simulation, for example, the discovery of the allosteric site of TEM-1β-lactamase.31 In conclusion, the discovery of hidden allosteric sites has greatly expanded the range of available drug targets, and these calculations have enabled us to predict hidden allosteric sites with greater accuracy, providing an opportunity for the discovery of allosteric drugs.
In addition, some tool used online can also be used to identify allosteric sites. (1) Allosite (http://mdl.shsmu.edu.cn/AST): this is the first publicly available allosteric site recognition method established by Professor Zhang Jian’s research group of Shanghai Jiaotong University in 2013.36 This method discovers and validates new targeted allosteric on many proteins such as CDK2α.37 (2) GO model (http://www.ligbuilder.org/cavity/home.php): Professor Lai Luhua of Peking University established a coarse-gained two-state Go model to identify allosteric sites. This method predicted two potential allosteric sites in Escherichia coil phosphoglycerate lactate dehydrogenase.38 (3) PARS (http://bioinf.uab.cat/pars): Panjkovich and Daura developed a method to identify the location of allosteric sites by normal mode analysis. PARS has successfully identified a number of previously discovered allosteric sites.39 (4) SPACER (http://allostery.bii.a-star.edu.sg): Goncearenco et al developed a method of allosteric site recognition based on the combination of Monte Carlo simulation and NMR.40
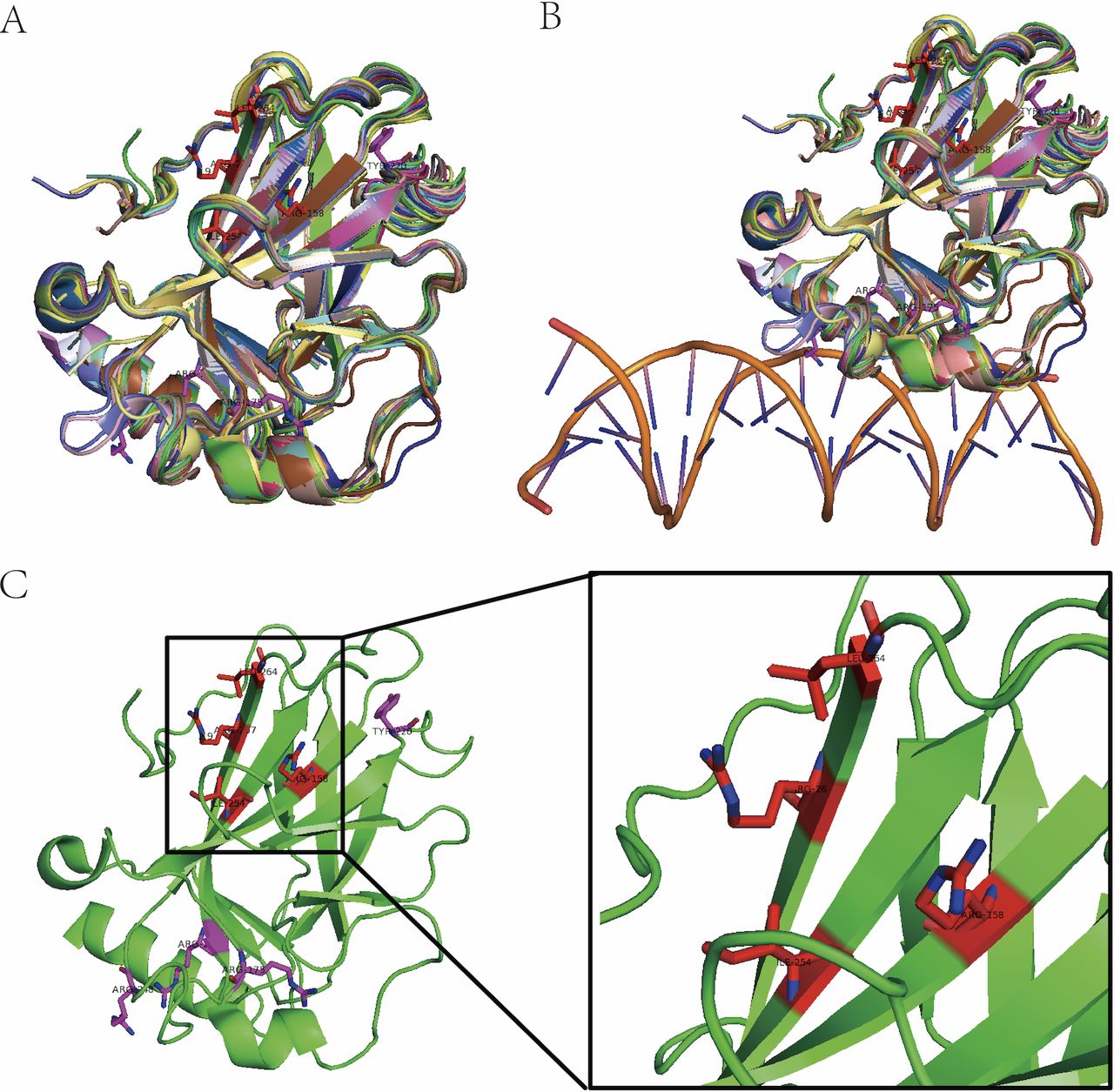
In the past few years, we have successfully used MD simulation to identify a potential allosteric pocket on DNA binding domain of p53 (figure 4). Based on this pocket, we have screened an allosteric lead compound both effective efficient in vitro and in vivo (unpublished data provided by Jingwei Jiang).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Identification of allosteric pocket on mutant p53. (A) Three-dimensional (3D) structural alignment of 43 p53 DNA binding domain. (B) DNA binding pocket is composed of fluctuating coli structures (double helix structure represents DNA). (C) Allosteric pocket (red amino acid residues) on p53(4lof) predicted by molecular dynamic simulation and hot spot mutant residues (purple).
Conclusion remark
In recent years, with the development of computational advancements and the unremitting efforts of scientists, they have improved innovative methods to overcome targets that are previously considered undruggable. At present, some lead compounds related to the undruggable targets have been developed. Some small molecules are already in clinical trials such as AMG-510 (a KRAS G12C inhibitor, Clinical Trial: NCT03600883). Soon, AI technology has also developed by leaps and bounds. Scientists have used computer technology to assist in the discovery of new drug targets and in search for hidden targets as well as undruggable targets, and have achieved certain positive results. Although AI cannot guarantee 100% accuracy just by computation, it still has many advantages compared with traditional methods in terms of cost and efficiency. This will also be the main trend in the future to discover potential allosteric sites on undruggable targets.
References
Footnotes
Contributors All authors wrote the manuscript. JJ provided guidance and modifications.
Funding This work is supported by NSFC (No. 81872892 and No. 2018ZX09735001-004), “Double First-Class” University project (No. CPU2018GY20 and No. CPU2018GY38).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.