Article Text

Abstract
There has been growing interest and insight into the histological composition of retrieved stroke emboli. One of the main focuses of the stroke clot analysis literature has been the implications of clot composition on mechanical thrombectomy procedures. However, the holy grail of clot analysis may not be in the field of clot–device interaction, but rather, in understanding mechanisms of fibrinolysis resistance. The mechanisms underlying the low response to fibrinolytic therapy, even with the newer, more powerful agents, remain poorly understood. While factors such as embolus size, location and collateral status influence alteplase delivery and recanalisation rates; compositional analyses focused on histological and ultrastructural characteristics offer unique insights into mechanisms of alteplase resistance. In this review, we strive to provide comprehensive review of current knowledge on clot composition and ultrastructural analyses that help explain resistance to fibrinolysis.
- stroke
- thrombectomy
- thrombolysis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Over the past two decades, there has been growing interest and insight into the histological composition of retrieved stroke emboli. Numerous groups have published instructive analyses of emboli from patients with acute ischaemic stroke (AIS) providing additional information regarding clot organisation, composition and ultrastructural analyses.1–9
One of the main focuses of the stroke clot analysis literature has been the implications of clot composition on mechanical thrombectomy procedures. There is a growing consensus in the literature that stiff, fibrin and platelet-rich clots are harder to retrieve with current devices than softer, red blood cell (RBC)-rich clots.1 10 11 In fact, the science has been so compelling that some companies have developed devices solely for the retrieval of these tough clots.12
However, the holy grail of clot analysis may not be in the field of clot–device interaction, but rather, in understanding mechanisms of fibrinolysis resistance. Among acute patients with AIS with large vessel occlusion (LVO), alteplase (tissue-type plasminogen activator) recanalised only 10% of occlusions while a newer fibrinolytic agent, tenecteplase (TNK), recanalised 22%.13 The mechanisms underlying the low response to fibrinolytic therapy, even with the newer, more powerful agents, remain poorly understood. While factors such as embolus size, location and collateral status influence alteplase delivery and recanalisation rates, compositional analyses focused on histological and ultrastructural characteristics offer unique insights into mechanisms of alteplase resistance. In this article, we will provide a comprehensive review of clot composition and ultrastructural analyses that help explain resistance to fibrinolysis and discuss potential future directions in the field of thrombolysis.
What are emboli from patients with acute ischaemic stroke composed of?
To date, several thousand of clots have been collected and analysed with emphasis primarily on histological composition. The dominant components studied in these analyses include RBCs, platelets, fibrin and white blood cells (WBCs). Histological analyses have demonstrated that emboli are highly heterogeneous. A review of our multicenter database of 1430 retrieved emboli from patients with acute ischaemic stroke found that 56% of emboli had RBCs as their dominant component while 21% had fibrin as their dominant component and 23% had platelets as their dominant component. Thus, the spectrum of clots can be classified as RBC-rich, fibrin-rich or platelet-rich clots.5 6 14
Review of the literature and large databases including those of the STRIP (Stroke Thromboembolism Registry of Imaging and Pathology) and RESTORE (REgiStry of cloT histOlogy and stRokE pathology) registries have found that there are two distinct structural components: (1) RBC-rich and fibrin-poor regions that have dense packing of RBCs within a network of thin fibrin strands with very few platelets and von Willebrand Factor (vWF) in between and (2) well-structured regions that are rich in platelets and fibrin and poor in RBCs and the platelet-rich areas have very dense fibrin structures and are rich in vWF with few RBCs.9 15 In general, these RBC-rich and platelet-rich regions are interspersed throughout the clot, while some clots are very homogeneous in their composition. Interestingly, a sizeable proportion of clots consist of core, which is RBC-rich and an outer crust of platelet-rich material.16
WBCs make up a minority of the cellular composition of emboli from patients with acute ischaemic stroke and comprise, on average, 2%–4% of clot area. The most common WBCs within emboli are neutrophils. These are predominantly found at the interface between the RBC and platelet-rich areas as well as within the platelet-rich zones. However, they are not commonly found in RBC-rich areas.15
One factor that is largely ignored in the ischaemic stroke literature is the concept of clot contraction. It is interesting that clots are often composed of distinct regions of RBC and platelet-rich areas, but why do different parts of the clots have such distinct architecture? Clot contraction likely plays a role in this process. It is thought that the contractile forces of platelets on fibrin can separate the RBC and platelet aggregates. This contractile process can compress RBCs as well to the form of polyhedrocytes.17 18 Clot contraction likely results in increased organisation, density and stiffness.19 Further work is needed to characterise this process in patients with ischaemic stroke.
Fibrin
Description and function
Fibrin, which is also known as Factor Ia, is a fibrous protein involved in haemostasis and is formed following cleavage of fibrinogen by thrombin resulting in polymerisation. Platelets are rich in thrombin receptors that bind serum thrombin molecules. Thus, when platelets start aggregating, they can turn local-soluble fibrinogen into fibrin.20 As the fibrin aggregates, it forms long strands of this tough insoluble protein that bind to the platelets. Factor XIII helps in completing the cross-linking of fibrin, so that it hardens and contracts.20
Identification in emboli
Histological studies of retrieved stroke emboli have demonstrated marked heterogeneity in the spatial distribution of fibrin and the proportion of fibrin in these clots (figure 1). Data from the STRIP clot registry show fibrin density ranges from 0.2% to 88% with the median fibrin content being around 27%. RBCs are generally the most well-represented component within emboli with clots from the STRIP registry having a median RBC composition of 43%. Fibrin networks in RBC-rich clots tend to be loose, which renders these clots somewhat more porous, thus allowing for improved delivery of fibrinolytic agents to their therapeutic target. Meanwhile, preclinical studies have shown that platelet and fibrin-rich clots are generally more resistant to fibrinolysis than RBC-rich clots.
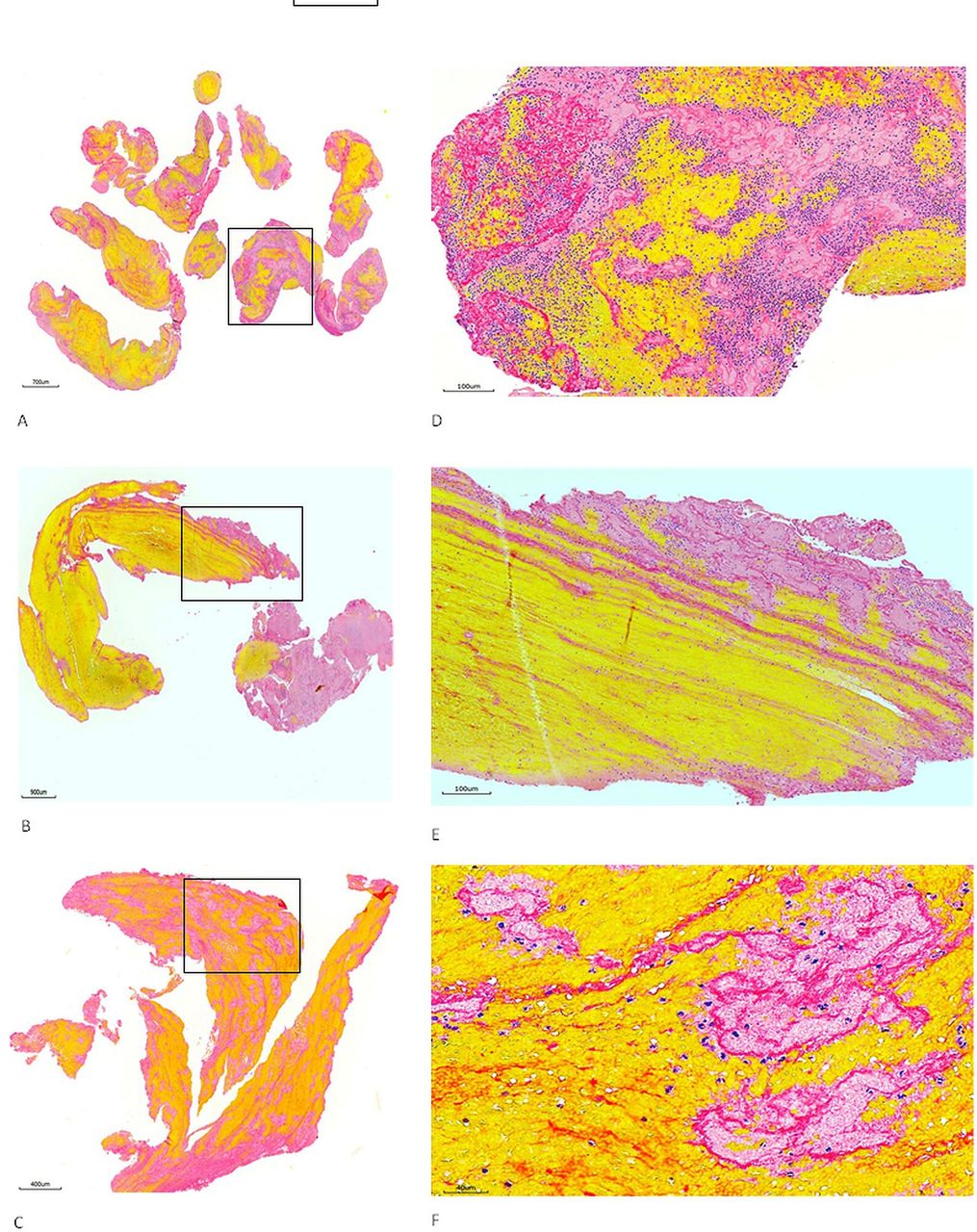
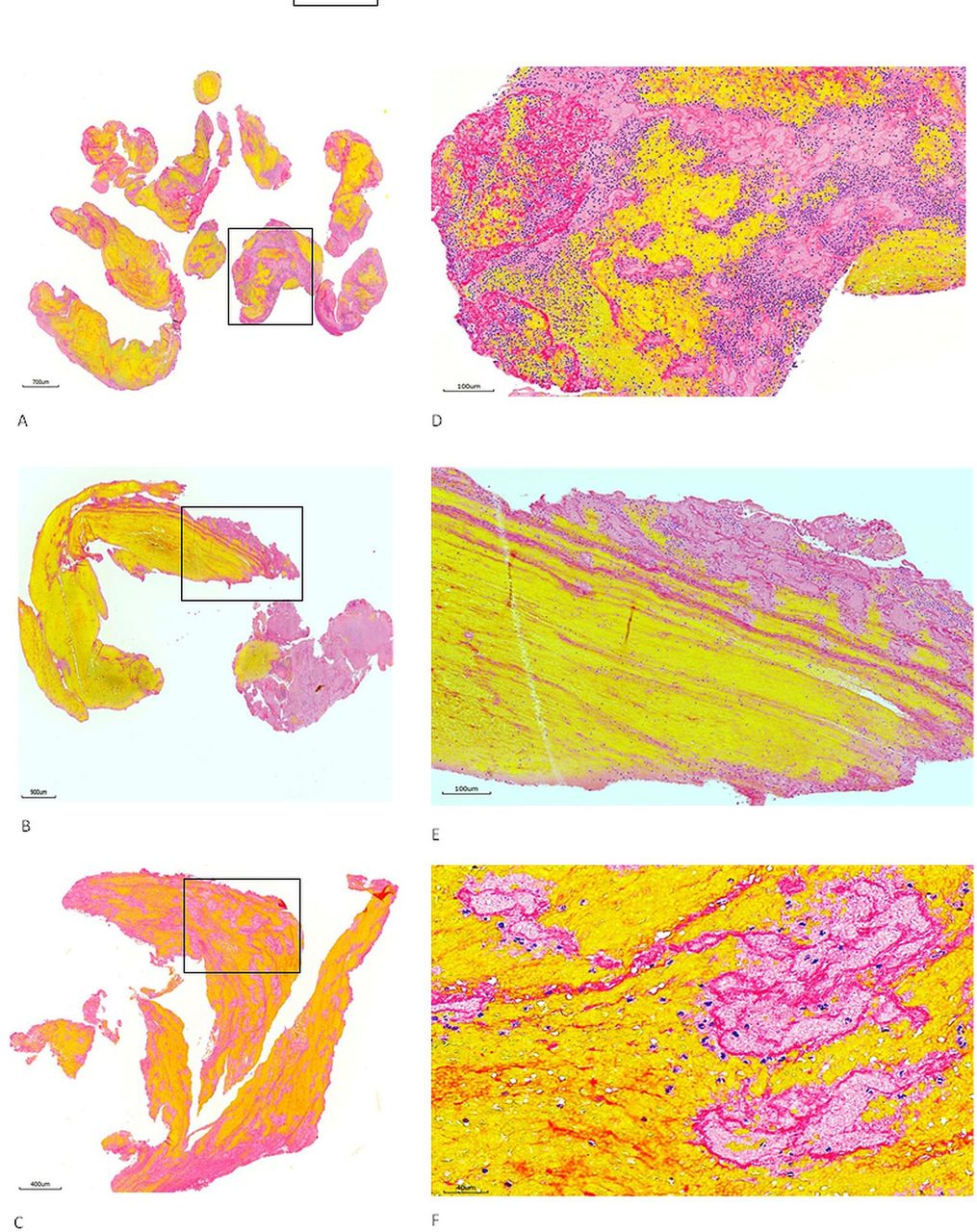
Heterogeneous feature of stroke emboli. At least two serial sections of thrombi retrieved from ischaemic stroke patients were stained with Martius scarlet blue to identify different component of thrombus. (A–C) Representative microphotographs taken with lower magnification from three different patients’ thrombi, showing the thrombus tissue consists of different component which is stained with different colour (yellow, pinkish, red and blue). (D–F) Microphotographs taken with higher magnification from the rectangular area in (A–C), showing red blood cells (yellow), fibrin strands (red/ intense pink), platelets (pinkish) and white blood cells (blue) all are present.
Fibrin heterogeneity
Fibrin structure and properties have been shown to be affected by numerous internal and external factors. Several studies have shown that genetic variants of fibrinogen and post-translational modifications could contribute to the heterogeneity of fibrin present in the clot. Mutations to fibrin gene can result in defective fibrinogen molecule, which is generally observed in dysfibrinogenemia.21 Genetic polymorphism of fibrinogen gene, cross-links of fibrin chains and oxidation, phosphorylation, methylation, glycosylation of amino acid moieties in fibrin may alter structure and function of fibrin.22–24
Therapeutic target
Since its approval by the FDA in 1996, alteplase has been the standard pharmacological agent for lysing clots in the setting of AIS. alteplase binds to fibrin within the clot and activates plasminogen within the clot. By cleaving plasminogen at its Arg561-Val562 peptide bond, alteplase creates plasmin, which is a fibrinolytic enzyme that cleaves the cross-links between the fibrin polymers. By cleaving the fibrin bonds within the clot, the clot will typically dissolve. Alteplase is inhibited by plasminogen activator inhibitor 1 (PAI-1), which binds to the drug and forms an inactive complex. Fibrinolysis by plasmin is short lived due to the presence of plasmin inhibitors, which inactivate plasmin.
More recently, there has been growing interest in TNK as an alternative to alteplase. TNK is a recombinant fibrin-specific plasminogen activator, which also binds to the fibrin component of the clot and converts clot-bound plasminogen to plasmin. One major difference between TNK and alteplase, however, is that TNK has higher fibrin specificity and is more resistant to inactivation by PAI-1.
As explained previously, the RBC-rich areas tend to be composed of loose fibrin networks while the platelet-rich areas are composed of dense fibrin networks rich in WBCs, vWF and, as we will see later, neutrophil extracellular traps (NETs). A number of previously reported studies have found that RBC-rich clots respond better to alteplase than platelet-rich clots.25–28 We hypothesise that this increased efficacy is due to improved delivery of alteplase into the loose meshwork of fibrin in the RBC-rich areas when compared with the denser fibrin structures copresented with platelet-rich regions.
Other factors affecting fibrinolysis
Several studies have acknowledged the importance of biophysical determinants of fibrinolysis.29 A key player in the inhibition of fibrinolysis is the activated factor XIIIa (FXIIIa), which catalyses the cross-linking of fibrin between residues in the γ-chains and α-chains of fibrin monomers within individual fibres. Although cross-linking has minor effects on the overall fibrin network morphology, it significantly decreases the extensibility and elasticity of individual fibres and, therefore, stabilises the clot and makes it more resistant to degradation by plasmin.30 Moreover, FXIIIa promotes RBC retention in clots by α-chain cross-linking. This adds to the contribution of platelet contractile force and RBC deformability to clot structure and may determine thrombus composition.31
Inhibition of the fibrinolytic system may occur at the level of plasminogen activation, mainly by PAI-1 and PAI-2. Fibrin degradation is additionally inhibited by thrombin-activatable fibrinolysis inhibitor (TAFI). FXIIIa does not only catalyse intramolecular cross-links within fibrin but also catalyses intermolecular cross-links between fibrin and other proteins including fibrinolytic inhibitors such as 2-antiplasmin (2AP), PAI-2 and TAFI and complement C3. In particular, the cross-linking of 2AP to fibrin has the strongest effect on fibrinolysis. The inhibition of clot lysis becomes stronger when clot retraction occurs and FXIIIa prevents α2AP being expelled from the clot.30
Platelets
Description and function
Platelets are nucleus-free cell fragments produced by bone marrow megakaryocytes. They function to preserve vessel wall health in healthy blood vessels and are a primary mediator of haemostasis in the setting of endothelial injury. When a vessel wall is injured or an atherosclerotic plaque ruptures, subendothelial collagen is exposed and platelets adhere to the collagen GPVI (glycoprotein VI) and integrin a2B1 receptors. This stimulates intracellular signalling mechanisms, which trigger platelet activation and promote clot formation. In the setting of atherosclerosis, however, platelet adhesion is more dependent on platelet GP1b receptor binding to vWF, which will be described further below.
Identification in emboli from patients with acute ischaemic stroke
Until recently, platelets, surprisingly, were ignored in the ischaemic stroke literature as major contributors to clot composition. Our group and others have been working on identifying platelets using CD42b immunohistochemical staining as well as Martius scarlet blue staining. Data from large clot registries have now confirmed, as suspected, that platelets represent a major structural component of stroke emboli. In the STRIP registry, platelets comprise, on average, 25%–30% of clot area.3 In general, it seems that platelets colocalise with vWF (figure 2A,B), which binds to platelets through GPIb (glycoprotein Ib) receptors. It is thought that the vWF GPIb binding results in platelets slowing down, thus, allowing for binding of GPVI and integrin a2B1 to arrest platelet movement.32 33 This leads to an intracellular signalling cascade, which allows the release of platelet agonists such as thromboxane A2, thrombin and ADP, which then triggers activation of GpIIbIIIa.34 Activated GpIIb/IIIa then binds to fibrinogen and vWF, which results in platelet–platelet cross-linking.34

Colocalisation of platelets and Von Willebrand Factor (vWF). Serial sections of thrombi retrieved from ischaemic stroke patients were stained with antibodies against CD42b and vWF to visualise the copresence of platelets and vWF. (A and B) Representative macrophotographs of serial sections from one thrombus stained for CD42b (A) and vWF (B), showing the distribution (red/pink) of platelets and vWF are same (Immunohistochemistry, original magnification ×4.0).
One interesting facet of platelet activation in clots is the process of contraction. Activated platelets can remodel clot fibrin networks and actually increase the fibrin density and increase clot stiffness.15 In fact, this process is thought to be responsible for the poorer revascularisation outcomes seen in platelet-rich clots. Platelets also play a role in activation of WBCs.3 Activated platelets release cytokines which then go on to activate WBCs and localise them to the clot. Studies have shown a strong spatial link between platelets and WBCs in emboli.3
Therapeutic target
Platelets represent a prime therapeutic target for thrombolysis in AIS. As mentioned above, much of the platelet–platelet cross-linking is primarily mediated via GpIIb/IIIa receptors. GpIIb/IIIa antagonists have been a mainstay of intravenous and intra-arterial antiplatelet therapy for well over a decade in helping to manage and prevent thrombotic complications during endovascular procedures.2 In fact, simple intravenous therapy with these agents is extremely effective in the management of complications such as in-stent thrombosis, while alteplase therapy is not, largely due to the fact that these thrombotic complications are primarily platelet mediated.2 Thus, it seems logical that if such therapy could be integrated with fibrolytic therapy, superior revascularisation outcomes could be achieved. There is currently a trial ongoing titled the Multi-arm Optimization of Stroke Thrombolysis trial comparing the efficacy of adjunctive thrombolysis with argatroban (a direct thrombin inhibitor), eptifibatide (a GpIIbIIIa inhibitor) and placebo is being compared (NCT03735979).
von Willebrand Factor
Description and function
vWF is increasingly recognised as an important factor in stroke clot pathology and pathophysiology. vWF is the largest circulating plasma glycoprotein that mediates thrombosis by recruiting platelets to sites of vascular injury. Along with fibrinogen and fibrin, vWF links platelets together and helps in the stabilisation of clots. vWF is produced in the endothelium as an ultralarge protein within Weibel-Palade bodies, megakaryocytes (a-granules of platelets) and in the subendothelial connective tissues.
vWF’s primary function is to bind other proteins involved in haemostasis. Factor VIII is bound to vWF while inactive in the circulation and is released from vWF by the action of thrombin. Without vWF, factor VIII rapidly degrades. vWF also binds to collagen when it is exposed beneath endothelial cells. vWF binds to platelet GpIb receptors, mostly under high shear stress conditions as well as to multiple additional platelet receptors when activated by thrombin. Finally, vWF also self-associates, extending from the luminal surface, serving as a scaffold for thrombus formation.35
Identification in emboli from patients with acute ischaemic stroke
To date, there have only been a few studies examining the proportion and distribution of vWF in retrieved emboli. If vWF was to be found to be present in high quantities within these emboli, it could represent a potential therapeutic target. In a study of 91 emboli samples from the RESTORE registry, the mean vWF content was 30% with a high correlation between platelet and vWF levels and an inverse correlation between vWF and RBC levels. vWF was present in all emboli with amounts ranging from 5% to 50%. Furthermore, data from the RESTORE registry suggest a strong correlation between vWF and WBC density, suggesting a possible relationship between vWF and inflammation, something which will be discussed later.3 One interesting finding from STRIP is the fact that vWF seemed to be spatially distributed along the surface of the clot. One additional study on vWF in retrieved emboli found that relative vWF content was significantly higher in retrieved emboli treated with alteplase than those that were not treated with alteplase. This supports the idea that emboli resistant to fibrinolysis is rich in vWF.
Therapeutic target
vWF is cleaved by the metalloprotease ADAMTS13, which cleaves the vWF A2 domain. ADAMTS13 has been studied as a thrombolytic agent in both animal and benchtop models. In a mouse study in which the middle cerebral artery (MCA) was occluded by vWF-rich emboli, alteplase did not lyse the occlusions, but dose-dependent administration of ADAMTS13 dissolved the emboli with no systemic bleeding side effects.25 Another potential therapeutic strategy for targeting vWF is by blocking vWF–platelet interactions and reducing vWF monomer–monomer disulfide bonds using N-acetyle cysteine. More recently, preclinical studies of thromboembolic stroke demonstrated that an inhibitor targeting the A1 domain of vWF recanalised carotid artery occlusions better than alteplase.36Results such as these suggest that vWF could be a novel target for thrombolysis.
Neutrophil extracellular traps and WBC
Description and function
NETs have recently been implicated in thrombosis. NETs consistent of decondensed chromatic lined with granular and fibrous structures, which are released by neutrophils in order to kill foreign pathogens. One of the key components of NETs formation is citrullination of histones, which allows for decondensation of nuclear chromatin. Recently, NETs have been implicated in thrombosis as well as they have been found to provide a scaffold for platelets and RBCs and can influence the coagulation cascade.37 38
NETs have been shown to play a very important role in thrombosis where they can promote both arterial and venous clots. NETs assist in the localisation of procoagulant factors including tissue factor and factor XII. The decondensed DNA fibres within the extracellular space act as a scaffold for RBCs, platelets and prothrombotic molecules and play a putative role in thrombus stability. Cell-free DNA from NETs mediates thrombin generation in the Factor XII and Factor XI-dependent pathways. Histones induce thrombin generation by activating platelets through inducing fibrinogen-mediated platelet aggregation. NETs can also accelerate the extrinsic coagulation cascade by their interaction with tissue factor. In vitro experiments have shown that NET-induced tissue factor expression in endothelial cells accelerates clotting.37 38
Identification in emboli from patients with acute ischaemic stroke
Quantification of NETs in AIS emboli is lacking and most studies have quantified the WBC level. Interestingly, WBCs make up a minority of the cellular components of AIS emboli. The median proportion of WBCs in AIS emboli is about 3% with an IQR of just 2%–5%.38 However, WBCs, particularly neutrophils (CD66b positive), play an outsized role in thrombosis in the form of NETs, which can cover up to 60% of the surface area of AIS emboli.
NETs are revealed by the presence of prominent extracellular nucleic acid-rich areas located in neutrophil-rich zones. These areas stain highly positive for H3Cit (figure 3), the defining marker of NETs as well as neutrophil elastase. As mentioned above, there are growing data suggesting that NETs for a scaffold for platelets and RBCs. This is supported by work from a number of groups, which show that NETs are abundant at junctions between RBC-rich and platelet-rich regions within AIS emboli as well as along the surface of emboli.
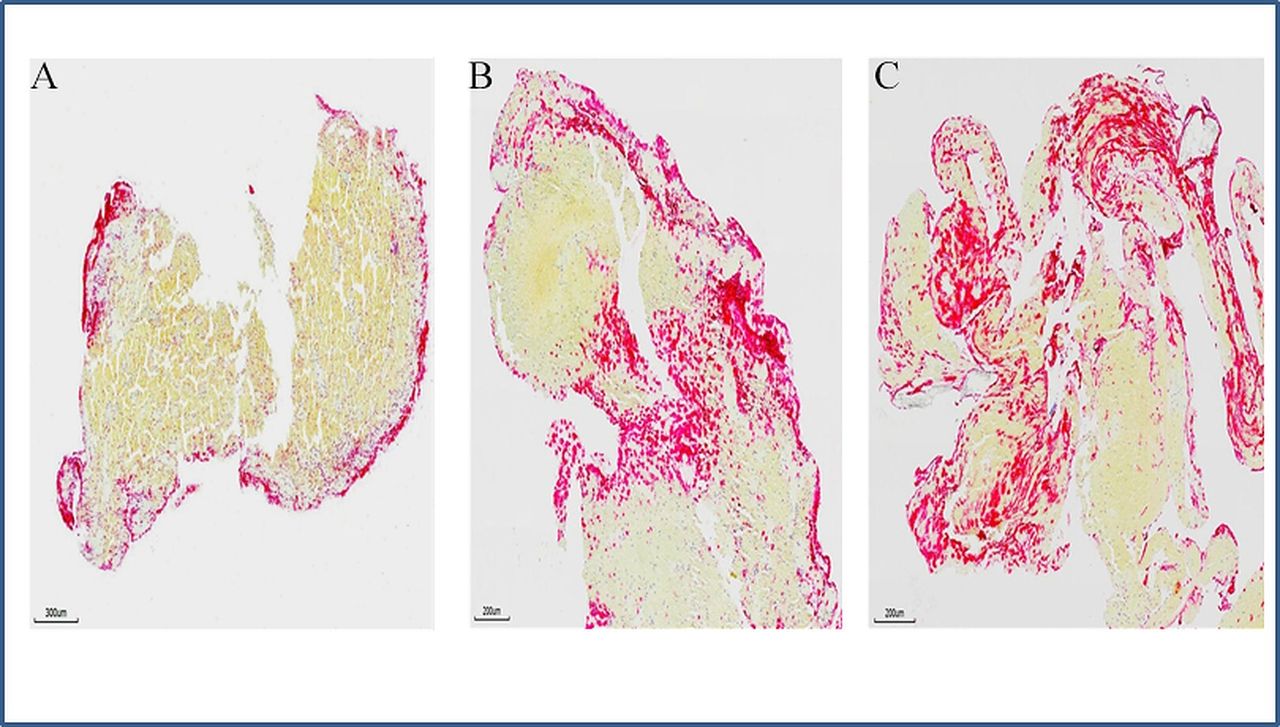
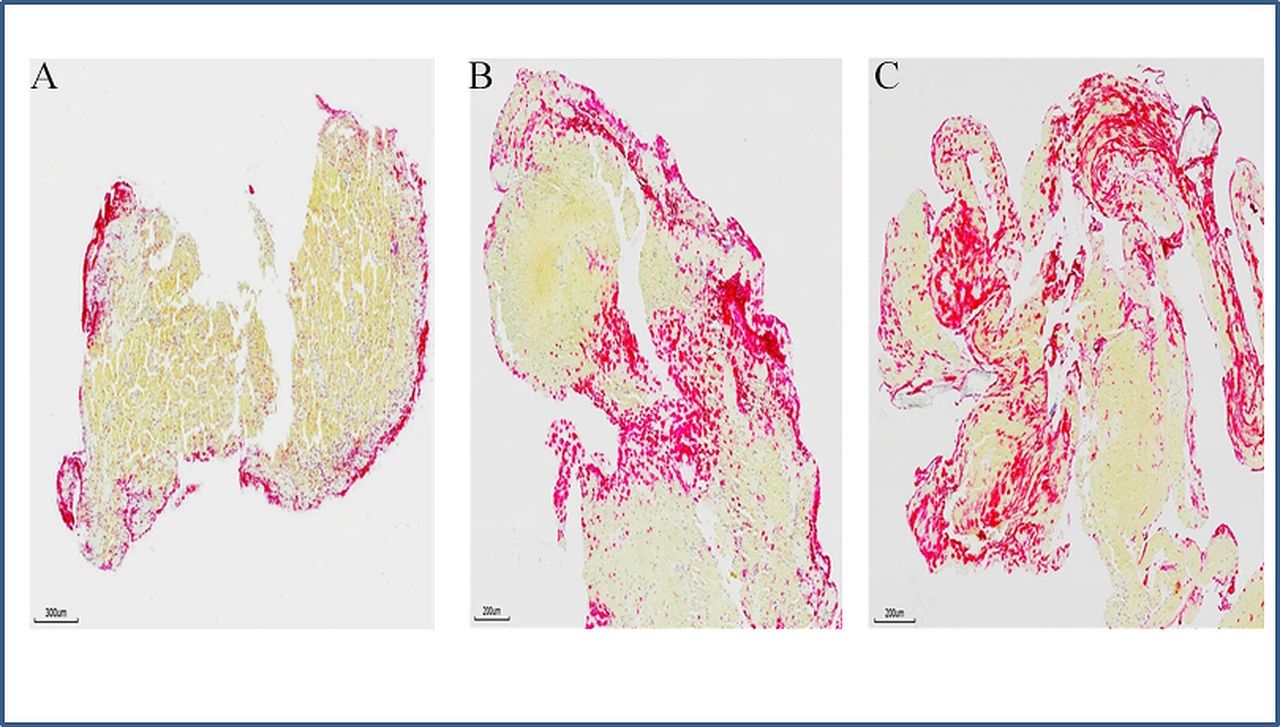
Neutrophil extracellular traps (NETs) in stroke emboli. Serial sections of thrombi retrieved from ischaemic stroke patients were stained with antibody against citrullinated histone H3 (H3Cit) to reveal the presence of NETs. (A–C) Representative microphotographs of three different patients’ thrombi stained for H3Cit (red), showing NETs is expressed along the surface of thrombus (A), at junctions between RBC-rich and platelet rich-regions (B and C). (Immunohistochemistry, original magnification ×3.7 (A), ×4.9 (B and C)).
NETs as a therapeutic target
Recent work has focused on the role of NETs as a potential therapeutic target in thrombolysis. NETs serve as a thrombotic scaffold contributing the clot stability and are thought to confer some degree of alteplase resistance. In fact, the addition of extracellular DNA and histones to fibrin has been shown to increase the thickness, stiffness and stability of the fibrin network; possibly making it harder to deliver alteplase to the interstices of the clot. High NET density has also been shown to make mechanical thrombectomy more difficult with increased number of passes and longer procedure times.38–40 However, DNA, the main component of NETs, is degraded by DNases, in particular, DNase 1 and DNase1-like 3.
In a seminal article by Jimenez-Alcazar et al, the authors found that DNase knockout mice produced vascular occlusions with NET-rich clots, resulting in significantly higher rates of end-organ damage than normal controls.41 In a study from De Meyer et al, addition of DNase 1 to alteplase resulted in significantly higher rates of ex vivo dissolution of emboli freshly retrieved from stroke patients. DNase 1 has also been shown to improve thrombolytic activity for coronary thrombi as well.
NETs–vWF–platelet axis
There is growing evidence linking NETs, platelets and vWF in a prothrombotic and proinflammatory axis. This axis is going to be very important to understand both for acute treatment of ischaemic stroke with thrombolysis and in primary and secondary prevention strategies; especially given the fact that NETs, platelets and vWF are the primary agents responsible for alteplase resistance of AIS emboli. vWF binds to pure DNA via electrostatic interactions, which, interestingly, can be blocked by heparin. The DNA binds to the same domain that the Gp1a receptors of platelets bind to as well. Neutrophils also adhere to vWF and can be recruited to the vessel wall via the vWF–DNA interaction. Neutrophil elastase, an abundant component of NETs, also binds to vWF via electrostatic interactions due to its positive charge. Thus, it is probable that vWF released from endothelial cells and platelets interact with NETs to promote progression of thrombosis and inflammation.
The vWF–NETs–platelet axis is further supported by histological studies of emboli. In a study by de Meyer et al, WBCs and extracellular DNA were present mainly in platelet-rich regions and at the boundary between platelet-rich and RBC-rich regions. It is thought that vWF serves as a mediator to bring NETs and fibrin into close proximity, thus facilitating the ability of NETs to modify the fibrin structure and increase local coagulation.38 Platelets play a key role in this axis as well and are thought to promote uncontrolled NETosis. Endothelial cell bound vWF promotes initial platelet adhesion by binding to platelet GPIba (glycoprotein Iba) receptors and activating platelets. These activated platelets promote neutrophil migration via toll-like receptors, which can promote the growth of clots.42 In fact, in one recently published study, platelet TLR4 was found to mediate NET formation in a mouse model of acute ischaemic stroke and ex vivo fresh platelet-rich emboli from patients were lysed by DNase-I.42
In summary, NETs and vWF recruit platelets and WBCs and, thus, promote coagulation. We believe that the NET–vWF interaction provides an extremely strong scaffold within emboli and facilitates interactions between platelets–WBCs–NETs and fibrin–vWF and NETs. Therefore, devising attempts to target the NET–vWFvplatelet axis could represent a viable therapeutic strategy for ischaemic stroke. A summary table is provided in table 1.
Summary table
Ultrastructural analysis of clot
Electron microscopy has opened the door to new insights into the ultrastructural characteristics of stroke emboli and their implications for fibrinolysis resistance. In one recently published study, Di Meglio et al analysed nearly 200 emboli using immunohistochemistry and scanning electron microscopy and found that stroke emboli shared a common structural feature comprising of an outer shell composed of densely packed fibrin, vWF and aggregated platelets.16 In vitro experiments confirmed that platelets were essential to the formation of this outer shell and this outer shell was found to decrease the efficacy of alteplase (figure 4).

Representative scanning electron microscopy (SEM) images of an acute ischaemic stroke thrombus. (A) Arrows correspond to the locations analysed by SEM. The surface of the clot shows multiple folds and ridges. (B) Distinct thrombotic components are not discernible on the surface that appears smooth. (C) High magnification of the surface suggestive of dense integration of components and advanced organisation. Only a few red blood cells are evident. (D) The rift on the surface shows the presence of a dense outer shell (arrow) and a different structure of the interior of the clot (asterisk). (E) Magnified area of the interior of the clot displays numerous individual polyhedrocytes and some distinct fibrin strands suggesting an immature structure. (F) The cross-section exposes the thrombus core which is detailed in (G). (H) Identifiable thrombus components indicate a limited maturity and incomplete integration. Scale bar=10 µm (C, E, H), 100 µm (B, D, G), 200 µm (F) and 500 µm (A).
Another recently published study of 3D electron microscopy (3DEM) in AIS emboli was also able to provide some novel insights into the ultrastructure of AIS emboli.8 In this particular analysis, 3DEM showed that RBC-rich areas consisted of polyhedrocytes, a morphological marker of clot contraction, which, as described above, is a common process, resulting from contractile platelets pulling on fibrin. Furthermore, the 3DEM data showed that WBCs were concentrated mainly in the fibrin-rich and platelet-rich areas and occasionally at the interface between RBC-rich and platelet-rich areas, lending further support to the potential role of NETs as a mechanism of fibrinolysis resistance. 3DEM was also able to show how alteplase resulted in thinning of fibrin fibres and disruption of fibrin fibre organisation (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Three-dimensional scanning electron microscopy analysis in three representative cases. Volume renderings assembled from serial block-face imaging highlight detailed ultrastructural organisation and characteristics of individual components for each clot (red blood cells (RBC)=yellow arrows, fibrin=red arrows, white blood cells=blue arrow). (A) Clot area with tightly packed RBC as polyhedrocytes intermixed with a limited volume of thin fibrin fibres. (B) Clot area with mixed composition consisting of both packed polyhedrocytes and dense network of thick fibrin fibres. (C) Fibrin-rich area containing dense fibrin masses with sparse polyhedrocytes.
Recently, many groups have explored the role of transmission electron microscopy in evaluating the efficacy of thrombolysis. For example, in a study evaluating alteplase versus TNK for in vitro clots, Fruhwald et al found that clots treated with TNK had significant reductions in fibrin fibre density than those treated with alteplase.43
Future directions
Over the past several decades, the main focus for stroke thrombolysis has been on fibrinolysis. However, if there is any chance for the field of pharmacological-based revascularisation to progress, the knowledge of clot composition, organisation and physical characteristics that we have gained from the multitudes of retrieved AIS emboli needs to be applied. First and foremost, more in vitro and animal studies studying the basic biologic mechanism of resistance to thrombolysis needs to be conducted, specifically targeting platelets, NETs and vWF. This requires the production of physiologically representative AIS clot analogues that are rich in these components and have similar structural organisation (ie, shell of alteplase resistant material) and physical characteristics and the application of thrombolytic agents in in vitro models, which replicate the haemodynamics of a vascular occlusion. This last point is especially true as one of the main challenges in thrombolysis is how to deliver drug to a blood vessel that has no flow.
Studies examining the effects of various novel thrombolytic agents on the ultrastructure of clots would also be useful, simply to understand what the exact mechanisms are for success and failure of these agents are. In addition, studies on mechanically altering the clot ultrastructure (such as sonothrombolysis) to enhance diffusion of thrombolytic agents into the clot can also be useful.
Conclusions
We are only starting to understand the complex biological and structural characteristics of thromboembolisms causing LVO stroke. Based on what we now know from our experience of retrieved emboli, these clots are composed of a complex mixture of RBCs, fibrin, WBCs, vWF, NETs and platelets with the fibrinolytic-resistant components (ie, vWF, NETs and platelets) located predominantly at the surface of the clot. Further experiments are needed to design and test novel thrombolytics that target not just fibrin, but these other components. Also, more research is needed to better understand the ultrastructural characteristics of stroke emboli in order for us to better understand mechanisms of thrombolysis resistance.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors WB, OMM, DD, DFK, LS, YL, SMN, RGN, MA and RK made substantial contributions to the conception or design of the work or the acquisition, analysis or interpretation of data for the work; and drafting of the work or revising it critically for important intellectual content. All authors provided final approval of the version to be published. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding This work was supported by the National Institutes of Health grant number (R01 NS105853).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.