Article Text

Abstract
Spontaneous intracerebral haemorrhage (ICH) is a devastating type of stroke with high mortality and morbidity and for which no effective treatments are available to date. Much experimental and clinical research have been performed to explore its mechanisms regard the subsequent inflammatory cascade and to seek the potential therapeutic strategies. The aim of this review is to discuss insights from clinical settings that have led to the development of numerous animal models of ICH. Some of the current and future challenges for clinicians to understand ICH are also surveyed.
- stroke
- inflammatory response
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction and definitions
Acute spontaneous intracerebral haemorrhage (ICH) (non-traumatic) affects approximately 2 million people each year in the world, and it is the most serious and least treatable form of stroke.1 Stroke was also the second most common cause of disability-adjusted life years, and according to a report from the Global Burden of Disease 2016 Lifetime Risk of Stroke Collaborators, the estimated global lifetime risk of stroke in 2016 for those aged 25 years or older was 24.9%.2
ICH indicates that blood has deposited in the brain parenchyma3 and may extend into the ventricles. Blood components, including leucocytes, haemoglobin, thrombin, plasmin, complement, plasma and fibrin degradation products appear in the brain tissues.4 An inflammatory response and brain cell death take place subsequently, which may involve enzyme activation, cytokine release, leucocyte migration and brain tissue breakdown and repair.5 Forty per cent of patients die within the first 30 days,6 the mortality rate at the first month is 43%–51%, and the survivors have irreversible consequences dependent on the injured location.6 The most common neurological deficit is hemiplegia or anaesthesia.7 There can be dysphasia,8 cognitive deficits,9 emotional difficulties,10 daily living problems and pain.11 In addition, numbness or tingling is also a common deficit. ICH in the brain stem may influence vision, swallowing, breathing, balance and consciousness.12
The amount of research on ICH lacks that of ischaemic stroke. The mechanisms of delayed clinical deterioration after ICH still remain unclear. This review is mainly focused on summing ICH occurring in the clinical setting and in different animal models, and describes progress in pathophysiology of brain damage after ICH.
ICH in the clinical setting
The worldwide annual incidence of spontaneous ICH is 12–35 per 100 000 population, which accounts for approximately 15% of cerebral strokes; it has a higher mortality rate compared with that of cerebral ischaemia.13 The major high-risk factors for ICH are an elderly population,14 male sex,15 current smoking,16 excessive alcohol consumption,17 low total cholesterol level,18 long sleep duration,19 illicit drug use,20 Asians ethnic origin21 and genetic factors.22 Other clinical disease can cause ICH, including hypertension,23 coagulopathy,24 cerebral amyloid angiopathy (CAA),25 cerebral tumours,26 intracranial arterial aneurysm,27 vascular anomalies,28 brain trauma,29 premature birth,30 haemorrhagic conversion of stroke,31 posterior reversible encephalopathy syndrome,32 vasculitis,33 infective endocarditis,34 dural arteriovenous fistula,35 brain arteriovenous malformation,36 cavernous malformation37 and intracranial venous thrombosis38 (figure 1). ICH associated with hypertension remains the most common form of ICH.23 The importance of CAA is growing due to ageing population.25 Besides, drug-related ICH, particularly anticoagulants, is also a major cause of ICH.39
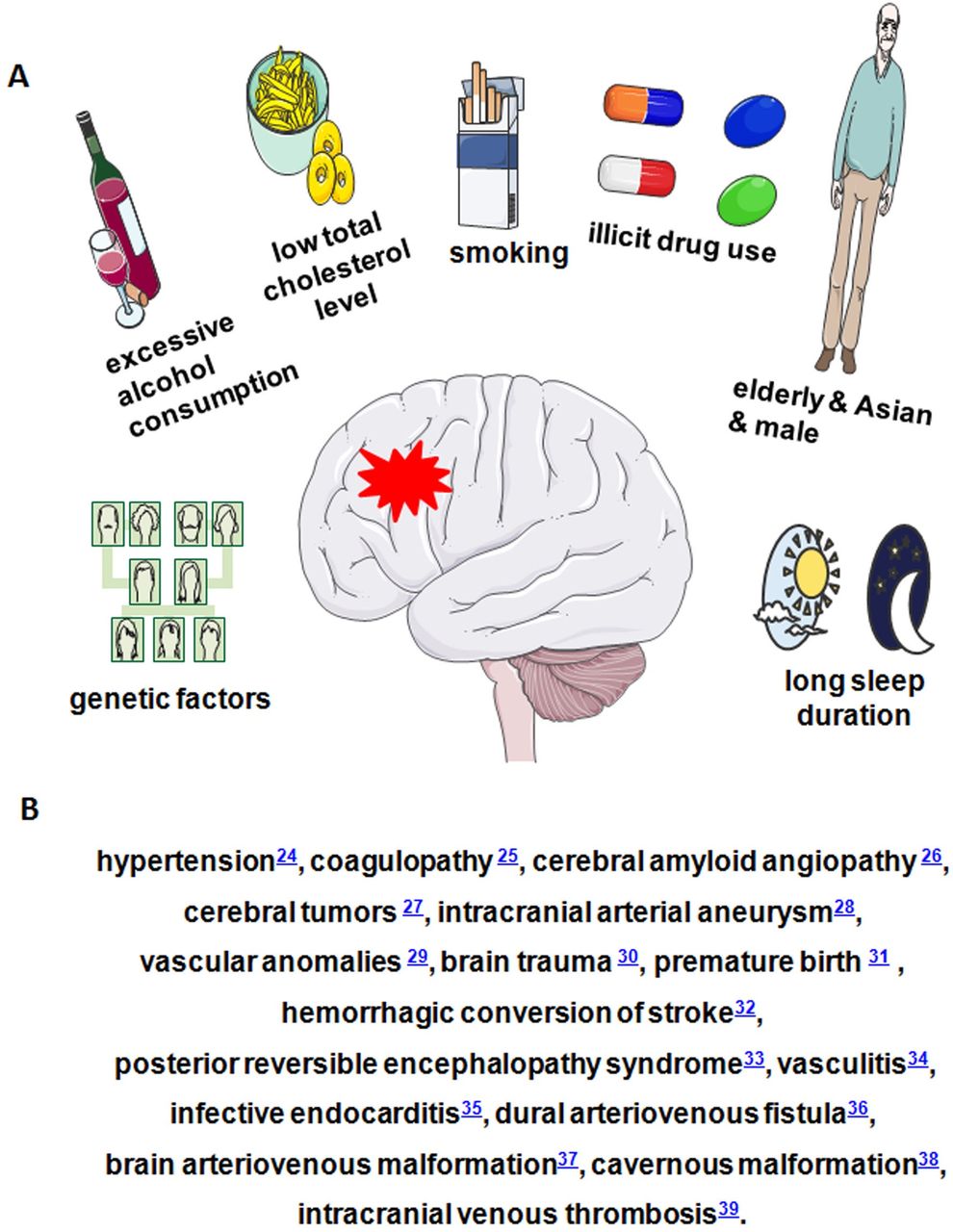
Risk factors of ICH. (A) Risk factors of ICH. (B) Some clinical diseases that can cause ICH. ICH, intracerebral haemorrhage.
Effective treatment for ICH is still scarce.40 However, clinical therapeutic strategies includes medication and surgery.41 Drug therapy is the most common treatment for ICH. This includes prevention of ICH based on treating an individual’s underlying risk factors, for example, control of hypertension.42 Hyperglycaemia in diabetics is common after stroke; managing glucose level may reduce the stroke size.43 Oxygen is given as needed. Surgery can be used to prevent ICH by repairing vascular damage or malformations in and around the brain, or to treat acute ICH by evacuating the haematoma; however, the benefit of surgical treatment is still controversial44 due to very few controlled randomised trials. Rehabilitation may help overcome disabilities that result from ICH damage.45
Animal models of ICH
Animal models of ICH may help us to understand its pathogenesis and explore preventive or therapeutic approaches. Experimental ICH models have been studied in several species including mouse,46 rat,47–49 rabbit,50 51 cat,52 pig53 and primate.54 The ICH model should be selected carefully to fit study aims (table 1).
Animal models of ICH
Microballoon insertion models
An acute expanding lesion model using a mechanical microballoon to simulate the space-occupying effect of ICH was developed by Sinar in the adult rat in 1987 (although this model lacks the effects of blood components).55 The microballoon system consists of an embolisation balloon mounted on a 20-gauge venous cannula using its own previously blunted guide. The microballoon is then inflated with saline in a syringe. After balloon inflation in the caudate nucleus of rats, intracranial pressure (ICP) increases significantly and cerebral blood flow (CBF) decreases subsequently in ipsilateral frontal cortex and caudate nucleus.56 Microballoons (25 mL and 50 mL in volume) that mimic lesion size in man cause little change in ICP. However, a larger volume (100 mL) increases ICP.57 The volume of ischaemic damage in the ipsilateral caudate nucleus for inflation group was reported to be 10-fold more than that for sham-treated group.55 Compared with transient inflation groups, amounts of injured neurons in permanent groups was significantly higher.58 Deflation of balloon after 10 min was shown to improve clinical outcome and reduced CBF abnormalities in rats.59 Therefore, to defend the development of irreversible neurological deficits and death in this model, evacuation of expanding haematoma-like mass is necessary within a proper time window. Similarly, a microballoon inserted into the ventral posterolateral nucleus of the thalamus in cat caused a rapid reduction in CBF following gradual balloon inflation.60
Autologous whole blood injection models
Anatomically localised haematomas may be realised by this method without artificial agents.48 49 61 Except for needle insertion, autologous blood injection may closely mimic clinical ICH. The autologous blood extracted from tail or femoral artery of animal is directly injected into particular brain regions. Several studies employ this model to survey brain injury mechanisms. Hydrocephalus, cell death, inflammation and behavioural disorders may be induced by autologous blood injection in rats.47–49 62–64 Studies in the dog have shown that despite a prominent increase in ICP and mean arterial pressure after ICH, ischaemic penumbra in the first 5 hours after ICH was not demonstrated.65 Increased ICP as well as compromised CBF and metabolism following ICH have been shown66 in the cats, rabbits, monkeys and pigs. Pigs have been frequently studied for clot evacuation.67 68 For instance, a tPA (tissue Plasminogen Activator)-induced clot lysis study showed that reduction in clot size was significantly greater than mechanical aspiration alone. In the rabbit model,69 urokinase treated animals showed 86% of clot lysis compared with injection of saline into clot (23%). Effects of blood components, including leucocyte fractions, erythrocytes, plasma, serum, thrombin and plasmin, were demonstrated separately in rats.49 70 Leucocytes, activated leucocytes, thrombin and plasminogen caused brain oedema, inflammation and brain cell death when they were injected into the brain.47 Components of the coagulation system can modulate inflammation.71 Activation of the complement system72 and injections of haemoglobin as well as erythrocytes into the brain may lead to brain oedema.61 70
Collagenase animal model of ICH
This model was developed by Rosenberg’s group. Bacterial collagenases, which may destroy capillary basal lamina, are injected into basal ganglia to induce ICH.73 74 Reproducible haemorrhage without significant blood leakage along the needle track mimics spontaneous ICH. Following ICH induced by collagenase in rats, behavioural improvement is rapid but incomplete at 3 weeks, accompanied by resolution of the oedema.62 This model is also used to study treatment following ICH.73–77 Compared with autologous blood injection model and venous haemorrhage model by avulsion of cerebral surface vessels, collagenase model introduces exogenous protein that may cause more inflammatory reactions.64 Addition of heparin to collagenase injection enhances the inflammation in rat brain.62 From an anatomical perspective, the extent of brain injury is more consistent for collagenase than other models. However, from a biological perspective, it is the most artificial. In addition, compared with other models, inflammation and cell death begin earlier. Collagenase induces a haematoma, and may cause cell damage directly and rapidly. Thus, the model has distinct differences from ICH in the clinical setting and the autologous bloodinjection model.
Thrombin model of ICH
Thrombin toxicity activates microglia and promotes cytokine production that causes neuroinflammation and cell death. Thrombin released from haematoma is a main contributor to secondary brain damage in acute ICH.47 78 Intraventricular injection of thrombin causes significant hydrocephalus, ventricular wall damage and periventricular blood–brain barrier (BBB) disruption.79 Intrastriatal thrombin injection that impairs neurogenesis and spatial memory function is partly mediated by inflammation, which is characterised by the activation of CD68 positive microglia/macrophages.80 This model has been used to study the mechanisms of thrombin toxicity that cause neuroinflammation and cell death.78 80 A disadvantage of this model is that it provides minimal utility beyond thrombin toxicity research.
Hypertensive stroke models
Hypertension is the most common risk factor for ICH. Hypertension also induces changes in the walls of small vessels in the brain leading to rupture, which make the blood bleed into the brain parenchyma. To understand the effect of hypertension induced haemorrhage and to develop treatment for it, several animal models have been developed.81 Renovascular hypertension may be induced by renal artery constriction. By means of ring-shaped silver clips, roots of both renal arteries are constricted.82 The rate of stable hypertension was 100% and the incidence of spontaneous stroke including ICH and brain infarct was 61.8% at 40 weeks after renal artery constriction.82 Furthermore, the induced hypertension is not dependent on renin, brain angiotensin and perhaps circulating vasopressin.81 Stroke prone spontaneously hypertensive rats may also develop cerebral haemorrhage as well as cerebral infarct.83 The brain lesions in this model include old and fresh cerebral haemorrhage and infarcts with or without subarachnoid effusion. These models simulate hypertensive ICH in humans and offer the chance to study the mechanism of brain injury following hypertensive ICH. The disadvantage is that brain lesions are unpredictable with regard to size and location.
Models of neonatal periventricular/intraventricular haemorrhage
Periventricular/intraventricular haemorrhage (PVH/IVH) occurs most commonly in premature infants of 24–30 weeks gestation.30 The mechanisms of germinal matrix (GM) haemorrhage have been illustrated in immature animals, including cats, dogs, rabbits and sheep. Fluctuations in arterial and venous blood pressure can cause PVH.84 In prematurely born rabbits (27–30 days gestation), IVH may be induced by glycerol to create intracranial hypotension.85 86 In a newborn beagle model, injection of phenylephrine hydrochloride intravenously induces hypertension which can cause IVH.87 Intraventricular injection of blood in dog has been employed to explore the influence of acute ventricular expansion on adjacent blood flow patterns.88 Dog models may be employed to survey risk factors for PVH/IVH.84 Unlike those seen in humans, superficial foci of bleeding may be induced in neonatal hypoxia mouse model.89 However, these researches are related to physiological and anatomical characteristics that allow occurrence of PVH/IVH, but not the tissue reactions. By injection of autologous whole blood into periventricular tissue including GM and striatum, we developed a novel PVH/IVH model in newborn mice.46 Haematoma expanded into the ventricles in most mice, which mimics GM haemorrhage in humans at 24–28 weeks gestation age. Therefore, according to imaging research in premature human infants, this model mimics grade III/IV PVH/IVH.90 This model provides an opportunity to study mechanisms of cellular injury after PVH/IVH. By injection of blood into the ventricles of the 7-day-old rats, posthaemorrhagic hydrocephalus may be induced.91 It mimics the hydrocephalus following PVH/IVH in prematurely born infants.92
Other animal models of ICH
In addition to the above-mentioned ICH animal models, others have also been developed. Cortical vessel avulsion by tearing the pia can cause mixed brain damage including ischaemic and haemorrhagic.64 Cortical vessel avulsion causes ischaemic infarction and haemorrhage. Therefore, it is not a simple ischaemic stroke model,93 94 but an ischaemia and haemorrhagic mixed model just like traumatic cortical laceration. Additionally, haemorrhage related to shaking injury in the 6-day-old rats has been studied as a model of child abuse.95 Some forms of traumatic brain injury also cause bleeding into the brain parenchyma.96–98 None of the above-mentioned ICH models completely reproduce the brain injury response following human ICH. However, these models have significantly contributed to the overall knowledge of the pathophysiology of human ICH including oedema, inflammation, cell death, brain damage, compromised CBF and metabolism as well as pathogenesis.
Pathophysiology of brain damage after ICH
ICH causes brain damage through multiple mechanisms.
Mechanical injury of brain: Mechanical injury of brain tissue may be induced by the expanding haematoma,29 56 and mechanical and chemical factors may reduce local CBF around the haematoma.56 Raised ICP and distortion of the microvasculature contribute to oedema and secondary brain damage.56 As cerebral oedema develops, ICP increases and cerebral perfusion pressure declines.56 In this regard, ICH has similarities to ischaemic stroke particularly in the penumbra region that surrounds the haematoma.99 100 If the haematoma is large and secondary infarction follows, the surrounding tissue may become necrotic.101
Complex immune and inflammatory cascades: Thrombin and plasmin are potentially toxic in the first day following ICH.102 Local ischaemia, release of toxins by blood breakdown products, release of iron,103 proteolytic enzymes or inflammation involving chemokines, cytokines and leucocytes all contribute to delayed damage.47 62–64 70 75 102 104 105 Degenerating erythrocytes and fragmented nuclear debris may be observed after 24 hours. Two to three days after ICH, erythrocytes begin to break down, and haemoglobin is released. Hemosiderin is evident in macrophages as early as 3 days after the bleed.106 Iron-dependent formation of oxidising agents results in brain damage.70 107 Damaged brain cells, reactive microglia/macrophages and neutrophils produce reactive oxygen species (ROS) that cause brain cell damage following ICH.29 108 Moreover, chemotactic factors, including thrombin, are released from blood clot and damaged brain after ICH.109 The transit of leucocytes from blood vessels into the insulted brain may be prompted by thrombin.110 Neutrophil infiltration and reactive glial changes including astrocyte activation and microglia reaction in the brain adjacent to the haematoma are obvious at 2–3 days after ICH.108 111–113 Secondary brain damage may be caused by activated leukocytes through liberating cytokines, ROS, NO, matrix metalloproteinases (MMPs) and other proteases.114 115 Large clots degrade very slowly because the macrophage ingestion of debris takes place only in the periphery of haematoma. For months after clot resolution, residual hemosiderin and mineralisation may be detected along the haematoma cavity. In IVH, blood debris may obstruct the cerebral aqueduct and cause hydrocephalus.41 116 The transition of proteolytic enzymes from plasma into the brain parenchyma, including thrombin, plasmin and complement proteins, may exacerbate ICH injury.117 MMPs are proteolytic enzymes with relative specificity for components of the extracellular matrix. Following brain injury MMPs, such as MMP-3 and MMP-9, are produced by infiltrating inflammatory cells, microglia and astroglia.118 Plasmin can promote the activity of MMPs.119 120 MMPs may injure directly by processing death molecules (eg, FasL), disrupting myelin and perpetuating inflammation119 120 (figure 2). This could also occur after ischaemia because large molecular weight proteins, including plasminogen and prothrombin, may penetrate the BBB.121–124 They may contribute to brain oedema102 125 (eg, albumin), cellular necrosis (eg, thrombin and plasmin) and inflammation47 (eg, complement).
Beneficialfactors: The proinflammatory microglia/macrophages play an important role in the early stages after ICH. However, increasing evidence indicates that the regulatory microglia/macrophages with potential reparative and anti-inflammatory roles in the later phase of ICH can resorb haematoma and resolve oedema, contributing to improved white matter integrity, repair and functional recovery.126 127 A recent study provesastrocytic-derived humanin could act as a beneficial factor in promoting a phagocytic/reparative phenotype.128

{kind=link}
{kind=link}
In the earliest stage of ICH, the primary injury causes blood products (Fe2+, Hb, thrombin) to leak into the damage area to activate microglia/macrophages to express high levels of IL-6, IL-1β, TNFα, GM-CSF, INFγ, ROS, RNS, CCLs, HO-1 and MMPs. These changes extend the brain damage such as brain oedema, cell death, blood–brain barrier disruption and neurological deficits. CCLs, chemokines subfamilies; GM-CSF, granulocyte-macrophage colony stimulating factor; Hb, haemoglobin; HO-1, heme oxygenase-1; INFγ, interferon-γ; MMPs, matrix metalloproteinases; RNS, reactive nitrogen species; ROS, reactive oxygen species; TNFα, tumour necrosis factorα.
Conclusion
Despite great advances in ischaemia stroke, no prominent improvement in the morbidity and mortality after ICH have been realised. The current understanding of ICH is still limited, and the models do not completely mirror the human condition. Novel effective modelling is required to mimic spontaneous ICH in humans and allow for effective studies on mechanisms and treatment of haematoma expansion and secondary brain injury.
References
Footnotes
QB and ZS contributed equally.
Contributors QB and ZS wrote the first manuscript and QB drew the images; YL and RZ searched and organised the papers; VWY edited the manuscript; MX supervised the project.
Funding The authors acknowledge operating grant support from the National Natural Science Foundation of China (grants no: 81870942, 81471174 and 81520108011), National Key Research and Development ProgramProgramme of China (grant no: 2018YFC1312200) and Innovation Scientists and Technicians Troop Constructions Projects of Henan Province of China (for MX); and from the Canadian Institutes of Health Sciences (VWY).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.