Article Text

Abstract
Background and purpose Data on the efficacy and safety of alteplase for acute ischaemic stroke (AIS) administered 3–4.5 hours after the onset of stroke symptoms in Chinese patients is limited. We sought to determine whether AIS patients would benefit from thrombolysis with alteplase between 3 and 4.5 hours after the onset of stroke symptoms in a prospective, multicentre, single-arm trial in China.
Materials and methods Eligible AIS patients were given 0.9 mg/kg alteplase intravenously. The primary efficacy endpoint was a favourable outcome at 3 months, defined as a score of 0 or 1 on the modified Rankin Scale. Thresholds for the primary efficacy endpoint were determined to be 40% based on the literature review. The primary safety endpoint was symptomatic intracranial haemorrhage (sICH) according to the European Cooperative Acute Stroke Study III (ECASS III) trial definition. Post hoc analysis between this study and the ECASS III trial were compared using the propensity score matching (PSM) method.
Results A total of 120 eligible AIS patients from 11 sites in China received thrombolysis therapy in this study. The median time from onset of symptoms to needle was 3 hours 54 min. The percentage of patients with a favourable outcome was 63.3% (95% CI 54.4 to 71.4), significantly higher than the predefined threshold (p<0.0001). Three patients (2.5%, 95% CI 0.5 to 7.1) had sICH, including two fatal sICH. Six patients died within 3 months after treatment. The post hoc PSM analysis showed a numerically higher rate of the primary efficacy endpoint in this study (63.3%) than the matched placebo arm (56.7%) in the ECASS III trial.
Conclusions Intravenous alteplase with a standard dose administered between 3 and 4.5 hours after onset of symptoms is effective and safe for Chinese AIS patients.
Trial registration number NCT02930837
- Stroke
- thrombolysis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The European Cooperative Acute Stroke Study (ECASS III) has demonstrated that intravenous alteplase administrated between 3 and 4.5 hours after the onset of symptoms significantly improved clinical outcomes in patients with acute ischaemic stroke (AIS).1 The subgroup analysis in ECASS III trial supported the use of alteplase up to 4.5 hours after the onset of stroke symptoms in various subgroups, including patients with older age, higher National Institutes of Health Stroke Scale (NIHSS) and longer time within the time window.2 In the meta-analysis or pooled analysis studies, intravenous alteplase was found effective and safe when delivered within 4.5 hours of stroke onset.3–5 The Safe Implementation of Treatment in Stroke-International Stroke Thrombolysis Registry (SITS-ISTR), a prospective observational study, demonstrated the effectiveness and safety for intravenous alteplase treated with the extended time window from 3 hours to within 4.5 hours after stroke onset in real world clinical setting.6–9 Hence, the American Stroke Association guideline recommended the use of alteplase in AIS patients in the 3 to 4.5 hours time window.10 11
The Safe Implementation of Thrombolysis in Stroke-Non-European Union World identified intravenous alteplase to be effective and safe in an Asian population when used within 3 hours of stroke onset.12 Several observational studies in Asian countries showed no significant difference in the efficacy and safety for thrombolysis treatment with intravenous alteplase in AIS between the 0–3 hours group and the 3–4.5 hours group.13–15 However, there have been no clinical trials for intravenous thrombolysis with alteplase in AIS patients between 3 and 4.5 hours in China, despite the ample evidence in the Western countries. Thrombolysis with alteplase is recommended for eligible AIS patients between 3 and 4.5 hour after onset of stroke symptoms by the national guideline in China since 2010.16 Under such circumstance, a head-to-head comparison between the alteplase and the placebo in a randomised controlled trial is impractical. We conducted a multicentre, prospective, single-arm, open-label study, in which the primary outcome was compared with the predefined threshold based on the data from the literature review.3–5 A post hoc analysis was conducted to compare the primary efficacy endpoint between this study and the propensity score matched arms from the ECASS III trial.
Materials and methods
Study design and patients
In this phase III, prospective, multicentre, open-label and single-arm study, eligible patients aged 18–80 years were enrolled in the study if they had received a clinical diagnosis of AIS, and were able to initiate thrombolytic therapy with alteplase within 3–4.5 hours after the onset of symptoms. A cerebral CT or MRI scan was required before alteplase administration to exclude intracranial haemorrhage (ICH). The inclusion and exclusion criteria are shown in online supplementary table I, which were similar to the ECASS III trial.1 Patients were enrolled from 11 sites in China between December 2016 and December 2017. All patients or their legally authorised representatives provided written informed consent before enrolment. The study was conducted in accordance with the guidelines of the International Council for Harmonization-Good Clinical Practice and the Declaration of Helsinki.
Supplemental material
Study drug treatments and concomitant medication
Eligible patients were given 0.9 mg of alteplase per kg, administered intravenously (with an upper limit of 90 mg), with 10% as a bolus and the remaining 90% infused continuously over 60 min. Administration of intravenous heparin, oral anticoagulants, antiplatelet agents (ie, aspirin), haemorrheologic agents, brain protective drugs (eg, calcium channel blockers) and volume expanders (eg, such as dextran and hydroxyethyl starch) were prohibited during the first 24 hours after completion of the study drug infusion. However, the use of low dose (≤10 000 IU) subcutaneous or equivalent doses of low-molecular-weight heparin for deep venous thrombosis prophylaxis was permitted during the first 24 hours.
Clinical and image evaluation
Neurological deficit was evaluated with the NIHSS17 18 at baseline, at 2 and 24 hours, as well as at 7, 30 and 90 days after the start of treatment. The disability was measured with modified Rankin Scale (mRS)19 at 30 and 90 days. Barthel Index Score20 was used to assess the recovery of neurological functions at 30 and 90 days. Glasgow Outcome Scale score21 was assessed the recovery at 90 days. All the measurements were performed by the investigators. CT or MRI scan was performed prior to alteplase treatment, between 22 and 36 hours after starting treatment, and additional scans were performed at the discretion of the investigator.
Outcome measures
The primary efficacy endpoint was a favourable outcome defined by a mRS of 0–1 at 3 months after onset of symptoms by face-to-face interview with patient. The secondary efficacy endpoint was a global outcome, measuring the percentage of patients who met the composite favourable outcomes (mRS score of 0–1, Barthel Index Score ≥95, NIHSS score of 0 or 1 and Glasgow Outcome Scale score of 1) at 3 months.
The primary safety endpoint was symptomatic ICH (sICH) according to the ECASS III criteria,1 including any apparent extravascular blood in the brain or within the cranium that was associated with clinical deterioration (defined by an increase in the NIHSS score of 4 or more points), or that led to death and was identified as the predominant cause of the neurological deterioration. The primary safety endpoint was evaluated by the data monitoring committee (DMC) consultants independently. Other safety endpoints included mortality rate within 3 months, death related to stroke or neurological causes, cerebral herniation and symptomatic cerebral oedema and other adverse events (AEs).
Statistical method
Sample size was considered both from efficacy and safety perspectives. For the primary efficacy endpoint, the percentage of patients with a favourable outcome was estimated from a historical review of intravenous thrombolytic therapy of alteplase within 3–4.5 hours after onset of symptoms. In the ECASS III study, the percentage of patients with a favourable outcome was 52.4%, and in the TIMS-China study as well as the SITS-ISTR study, those with favourable outcomes were 60.9% and 40.5%, respectively.3–5 Hence the predefined threshold for the primary efficacy endpoint was 40%. Assuming the percentage of patients with a favourable outcome in Chinese patients to be 55%, we calculated that about 120 patients in the single arm would be required to provide a power of 90% to rule out the percentage of patients with a favourable outcome of 40% or lower. For the safety evaluation, an assumed sICH rate of 2.4% (3 sICH events out of 120 patients), with an exact binomial 95% CI of 0.48% to 6.99% (confidence width of 6.51%) was considered as achieving a reasonable estimation precision.
Efficacy and safety endpoints were assessed in the treated set population which included all enrolled patients who received any dose of study drug of alteplase. For the primary endpoint, the percentage of patients with a favourable outcome at 3 months was calculated along with its 95% CI. A one-sample test was performed at two-sided significance level of 0.05. The rate of sICH within 3 months and its 95% CI was calculated for the primary safety endpoint. The other binary endpoints, the percentages and the CIs were also calculated accordingly.
A post hoc exploratory analysis was performed to compare the data of this study to the landmark ECASS III trial on the primary efficacy and safety endpoints as well as death, using Nearest Available Neighbour method for Propensity Score Matching (PSM)22: 120 patients each were matched to ECASS III trial alteplase and placebo arms. The number of patients was based on PSM method at a 1:1:1 ratio, with adjusted factors including age, gender, weight, baseline NIHSS score, blood pressure (systolic and diastolic), diabetes, hypertension, atrial flutter or fibrillation, history of stroke and time to treatment initiation.
Results
Patients disposition and baseline characteristics
Between 12 December 2016 and 11 December 2017, 120 were enrolled and received the alteplase treatment within 3–4.5 hours after onset of symptoms (online supplementary material 1). Baseline demographic and characteristics are detailed in table 1.
Demographic and baseline characteristics
The mean age of patients was 61.1 (±10.8) years, the majority being male (80.0%). The baseline median NIHSS score was 6.0 (range 1–21), with nearly half of patients (46.7%) scoring 4–9, and two patients (1.7%) scoring >20. The median time from onset of symptoms to needle was 3 hours 54 min. A total of 21.7% of patients received alteplase at 3.0–3.5 hours after onset of symptoms, 39.2% of patients each received alteplase within the time window of 3.5–4.0 hours and 4.0–4.5 hours, respectively. A total of 10.8% of patients experienced prior stroke. Antiplatelet treatment was delivered in 1.7% of the patients before thrombolysis with alteplase, and 2.5% within 24 hours after thrombolysis. Thirty patients (25.0%) received anticoagulation therapy, none of the anticoagulation therapy began within 24 hours after thrombolysis .
Efficacy outcomes
The primary endpoint and secondary endpoint, including the individual neurological and disability scores are shown in table 2. Seventy-six out of 120 patients (63.3%, 95% CI 54.4 to 71.4) achieved the primary endpoint of a favourable outcome (mRS 0–1) at 3 months after alteplase treatment. The percentage of patients with a favourable outcome was significantly higher than the predefined threshold of 40% (p<0.0001). For the secondary efficacy endpoint, 66 patients (55.0%) achieved the global outcome response (95% CI 46.1 to 63.6). There was no significant difference for the proportion of favourable outcome in the three groups based on the time of treatment: 3–3.5, 3.5–4 and 4–4.5 hours (online supplementary table II).
Supplemental material
Primary and secondary efficacy endpoints at 3 months
A total of 120 patients each were matched to ECASS III trial alteplase and placebo arms after PSM. PSM analysis showed that the percentage of patients with a favourable outcome in this trial was comparable to the matched ECASS III trial alteplase arm (63.3% vs 61.7%; OR 1.07, 95% CI 0.64 to 1.81), and demonstrated a numerical improvement compared with the matched placebo arm (63.3% vs 56.7%; OR 1.32, 95% CI 0.79 to 2.22; table 2). The mRS distribution is shown in figure 1.

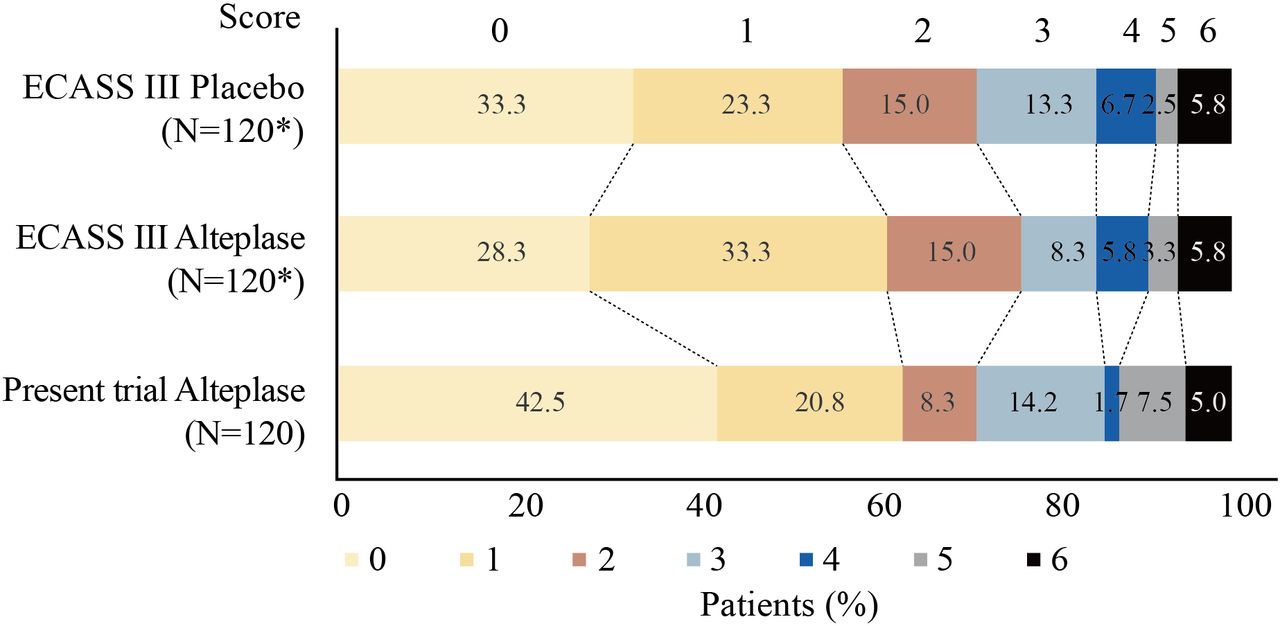{kind=link}
Comparation of the function outcome by the 3-month mRS in this trial to the matched ECASS III trial arms using PSM method at a 1:1:1 ratio. The proportin of favourable outcome was 63.3% in this trial, as compared with 61.7% in the matched ECASS III trial alteplase arm (OR 1.07, 95% CI 0.64 to 1.81) and 56.7% in the matched placebo arm (OR 1.32, 95% CI 0.79 to 2.22). ECASS III, European Cooperative Acute Stroke Study III; mRS,modified Rankin Scale; PSM, propensity score matching. * indicates the number of patients was based on Propensity Score Matching (PSM) method .
Safety outcomes
For the primary safety endpoint, 3 patients (2.5%, 95% CI 0.5 to 7.1) had sICH based on the independent DMC consultant’s evaluation (table 3). All of those sICH events occurred within 24 hours after starting alteplase treatment and were considered drug-related as assessed by the investigator; two resulted in death. All of those sICH occurred in three patients without heparin (online supplementary table III).
Supplemental material
Overall summary of AEs
A total of 7 out of 120 patients (5.8%) died, 4 between 2 and 7 days, 2 between 8 and 30 days, and 1 at 100 days. Five death cases were considered related to stroke or neurological causes. Cerebral herniation was reported in two patients. Symptomatic cerebral oedema was reported in three patients, all of whom were those experienced sICH. A total of 23 patients (19.2%) experienced serious AEs (SAEs). Drug-related AEs were reported in 15 patients (12.5%). All SAEs categorised by system organ class are shown in table 4.
SAEs within 3 months by system organ class
In the PSM analysis, two patients (1.7%) in the matched ECASS III trial alteplase arm and none in the matched placebo arm reported sICH. Seven patients (5.8%) died in each matched alteplase and placebo arms within 3 months, respectively.
Discussion
In this prospective, single-arm and multicentre phase III trial, alteplase was effective in Chinese AIS patients given within 3–4.5 hours after onset of symptoms. The majority of patients had a favourable outcome of mRS 0–1 (63.3%) at 3 months after thrombolytic therapy with alteplase, which was significantly higher than the predefined threshold of 40% (p<0.0001). The main result in this study was similar with the result in the ECASS III trial and supported the routine use of intravenous alteplase for AIS between 3 and 4.5 hours time window.
This study demonstrated the tendency for the efficacy of intravenous thrombolytic therapy with alteplase in AIS up to 4.5 hours after the stroke symptoms onset in the further PSM analysis. The efficacy of intravenous alteplase in AIS can be modified by factors such as the treatment delay, age and baseline stroke severity.6 Compared with the ECASS III trial and the meta-analysis, the patients in this study were younger with lower baseline NIHSS scores. The median age was 61 years old while the median baseline NIHSS was 6 in this study, compared with 65 years old and 9 in the alteplase group or 66 years old and 10 in placebo group in the ECASS III trial.1 The treatment delay were about 4 hours, similar among different trials. To balance these baseline characters, we compared the primary efficacy outcome in this single-arm study with the placebo arm in the ECASS III trial using PSM method. The percentage of patients with a favourable outcome (mRS 0–1) at 3 months in this study (63.3%) was numeric higher than the matched placebo arm (56.7%) in the ECASS III trial.
No new safety concern or difference in safety profile of alteplase was observed in this study. The 3 months sICH rate of this study (2.5%) was generally comparable to the whole ECASS III trial alteplase arm (2.4%; 10/418), although it was higher than that in the matched placebo group in the ECASS III trial.1 In TIMS-China study, no difference in sICH rates between TIMS-China and other previous trials was observed after using different definitions from different trials to evaluate the sICH.15 The overall mortality was comparable between this study (5.8%) and the matched placebo arm in the ECASS III trial.
This study is the first multicentre, prospective trial to evaluate the efficacy and safety of alteplase in Chinese patients with AIS given within 3–4.5 hours after onset of symptoms. There were some limitations in this study. First, this study is a single-arm study without a control arm which could result in difficulty with result interpretation. However, at the time of study initiation, alteplase was the most effective treatment of AIS and clinical evidence also supported the use of intravenous alteplase within 3–4.5 hours after onset of symptoms,11 12 thus it would not be ethical nor feasible to include a placebo arm in this study. Although a post hoc analysis using PSM method was performed to balance the important baseline characters including the age, stroke severity, treat time delay associated to the outcome, other potential factors may have an effect on the efficacy and the safety. At the same time, since the outcome measurement was not blinded, information bias may exist. Second, the threshold of 40% for the primary endpoint was defined based on the historical data from the ECASS III, TIMS-China and SITS-ISTR study, in which the lowest rate was chosen as the ineffective boundary.3–5
Conclusion
Administration of intravenous alteplase in Chinese AIS patients within 3–4.5 hours after onset of symptoms is effective and safe.
Acknowledgments
The authors met criteria for authorship as recommended by the International Committee of Medical Journal Editors, that is, were full responsibility for the scope, content and editorial decision, were involved at all stages of manuscript development, and approved the final version. The authors wish to thank Thierry Danays for his medical contributions, Na Hu and Erich Bluhmki for statistical contributions, as well as Changxia Shao for outputs deliverables. Thierry Danays, Na Hu, Erich Bluhmki and Changxia Shao are employees of Boehringer Ingelheim. The authors also wish to thank the members of independent data monitoring committee for their contribution, including Feng Chen, Yining Huang, Shaoshi Wang, Werner Hacke and Qiang Dong.
References
Footnotes
Contributors HZ: conception and study design, analysis of data, data acquisition, drafted the manuscript for intellectual content; YY, HC, CL, YC, F-DS, LY, XC, ZL, YL, SC and AX: data acquisition, revised manuscript for intellectual content; YQW: study design, analysis of data, revised manuscript for intellectual content; YS: study design, statistical analysis of data, revised manuscript for intellectual content; YJW: conception and study design, analysis of data, revised manuscript for intellectual content. All authors have approved the manuscript to be published. The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non exclusive for government employees) on a worldwide basis to the BMJ Publishing Group and its Licensees to permit this article (if accepted) to be published in Stroke and Vascular Neurology editions and any other BMJPGL products to exploit all subsidiary rights, as set out in our licence.
Funding The study was funded by Boehringer Ingelheim (China) Investment. The study was also funded by Clinical Research with Features for Application in the Capital (no. Z161100000516079) and the National Key Research and Development Plan (no.2017YFC1308204).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. To ensure independent interpretation of clinical study results, Boehringer Ingelheim granted all external authors access to all relevant material, including participant-level clinical study data, and relevant material as needed by them to fulfill their role and obligations as authors under the ICMJE criteria. Detailed requirement is provided in online supplementary material.