Article Text

Abstract
Intracerebral haemorrhage (ICH) is a devastating type of stroke with high mortality and morbidity. However, we have few options for ICH therapy and limited knowledge about post-ICH neuronal death and related mechanisms. In the aftermath of ICH, iron overload within the perihaematomal region can induce lethal reactive oxygen species (ROS) production and lipid peroxidation, which contribute to secondary brain injury. Indeed, iron chelation therapy has shown efficacy in preclinical ICH studies. Recently, an iron-dependent form of non-apoptotic cell death known as ferroptosis was identified. It is characterised by an accumulation of iron-induced lipid ROS, which leads to intracellular oxidative stress. The ROS cause damage to nucleic acids, proteins and lipid membranes, and eventually cell death. Recently, we and others discovered that ferroptosis does occur after haemorrhagic stroke in vitro and in vivo and contributes to neuronal death. Inhibition of ferroptosis is beneficial in several in vivo and in vitro ICH conditions. This minireview summarises current research on iron toxicity, lipid peroxidation and ferroptosis in the pathomechanisms of ICH, the underlying molecular mechanisms of ferroptosis and the potential for combined therapeutic strategies. Understanding the role of ferroptosis after ICH will provide a vital foundation for cell death-based ICH treatment and prevention.
- iron toxicity
- lipid peroxidation
- ferroptosis
- intracerebral hemorrhage
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Intracerebral haemorrhage (ICH) accounts for 10%–15% of all strokes. It is a devastating event that causes high mortality and morbidity, but it currently lacks effective therapies.1 ICH is estimated to affect over 1 million people worldwide each year, and the number is expected to rise substantially as the population ages. Most patients with ICH die or become severely disabled, and the mainstay for current therapy is supportive care. Consequently, only 20% of survivors have functional independence at 6 months.2 3 Primary brain damage develops in the first few hours after ICH as the formation and expansion of haematoma or oedema induce mass effects and elevations in intracranial pressure that lead to herniation and death. In the aftermath of ICH, iron accumulation within the perihaematomal region contributes to the secondary brain damage.4–12
ICH occurs when a weakened vessel ruptures and bleeds into the surrounding brain.6 13 The blood accumulates and compresses the surrounding brain tissue, causing tissue damage, inflammation and neuronal death.14–16 Preclinical and clinical studies suggest that toxins released from an intracerebral haematoma may contribute to brain damage after ICH.5–8 12–17 Haemoglobin (Hb)/haem is one putative neurotoxin. Hb, the most abundant protein in blood, is released from lysed red blood cells after ICH. It can be engulfed by microglia and infiltrating macrophages in the perihematomal zone and metabolised into ferrous/ferric iron, which induces the formation of lethal reactive oxygen species (ROS) and lipid peroxidation.8–12 Subsequently, excess ferrous iron is transported out of microglia, accumulates in neurons via the transferrin (Tf)–Tf receptor system and reacts with hydrogen peroxide (H2O2) via the Fenton reaction to form highly toxic hydroxyl radicals (•OH) (figure 1). These hydroxyl radicals attack DNA, proteins and lipid membranes, thereby disrupting cellular function.18 In our previous studies, we showed that iron toxicity contributes to ICH-induced early brain injury and that reducing iron accumulation with iron chelators (deferoxamine, 2,2′-dipyridyl, or VK-28) reduces grey and white matter injury and improves functional outcomes.8 9 11 Additionally, we were the first group to demonstrate that an iron-dependent form of non-apoptotic cell death known as ferroptosis occurs in a mouse model of ICH.10 Therefore, patients with ICH might benefit from a reduction in Hb/iron-induced toxicity or direct neuronal rescue.6 12

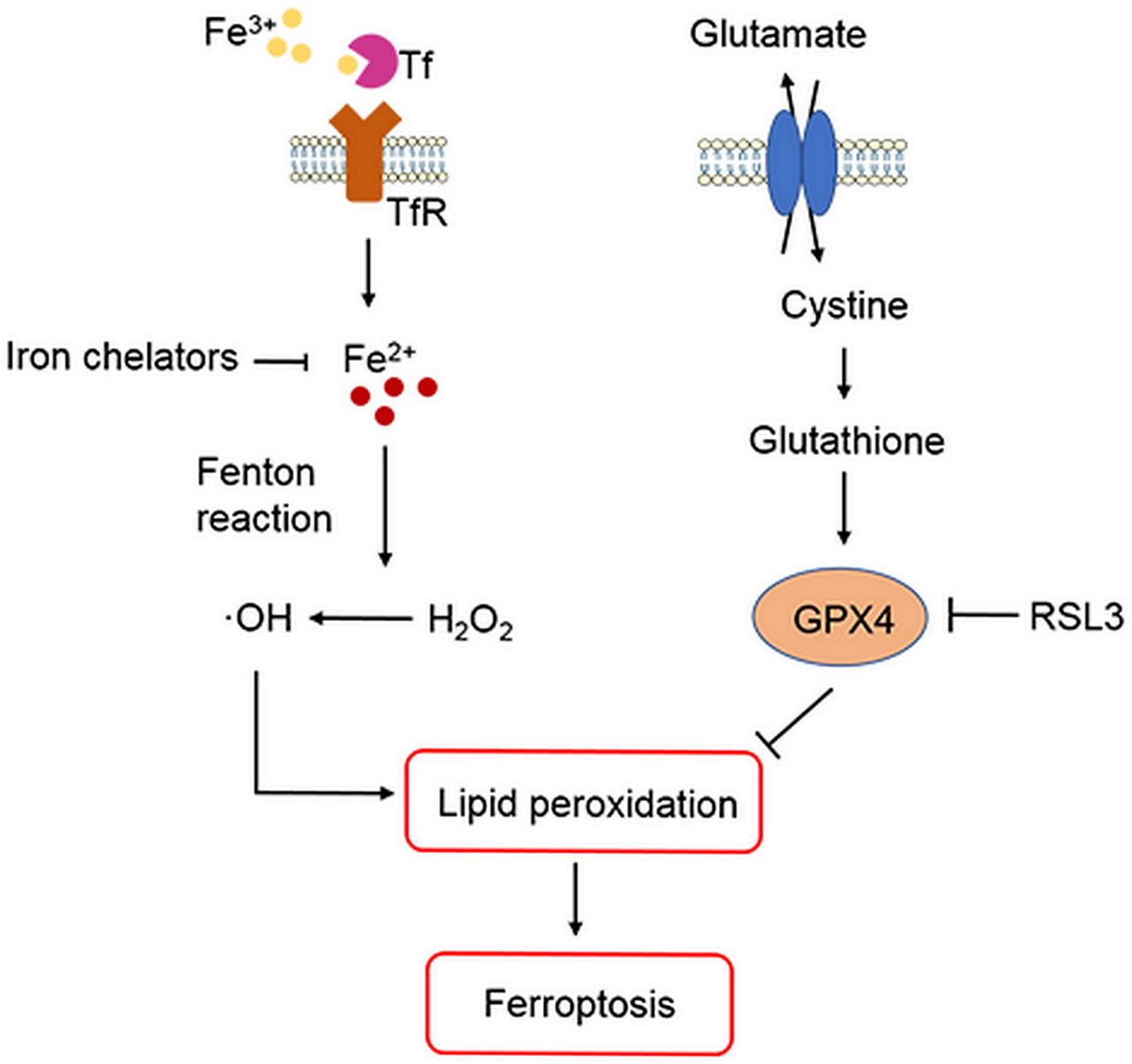{kind=link}
Role of iron, glutathione and lipid peroxidation in ferroptosis. Arrows indicate promotion; blunt-ended lines indicate inhibition. GPX4, glutathione peroxidase 4; H2O2, hydrogen peroxide; •OH , hydroxyl radicals; RSL3, Ras-selective lethal 3; Tf, transferrin; TfR, transferrin receptor.
Researchers have identified various forms of cell death after ICH, including apoptosis19–24 and necrosis20 25 in humans and experimental animals, and autophagic cell death26 27 in animal models. Whereas mitochondria are usually swollen in each of these forms of cell death, ferroptotic cells exhibit mitochondrial shrinkage. In their 2016 review, Xie et al 28 provide direct comparisons of the morphological, biochemical and immune features of ferroptosis, apoptosis, necrosis and autophagy. Inhibition of apoptosis, necrosis and autophagy improve outcomes in ICH animal models.22 25 27 However, caspase inhibitors have failed to inhibit neuronal death induced by Hb,29 and multiple forms of cell death may contribute collectively to neuronal death after ICH. Indeed, Zhu et al 25 showed that necrosis occurs earlier than apoptosis but that both contribute to cell death after ICH. Others have reported that both apoptosis and necrosis are present in brain sections from patients with ICH.20 24 These results prompted us to investigate whether ferroptotic cell death occurs after ICH. In a recent study, ferroptosis was identified in cancer cells and in organotypic hippocampal slice cultures (OHSCs) exposed to glutamate.30 Ferroptosis is distinguishable from other forms of regulated cell death because it does not require caspases (mediators of apoptosis and pyroptosis), ATP depletion or mitochondrial ROS generation (mediators of necroptosis), Bax/Bak (essential mediators of mitochondrial outer membrane permeabilization), or elevations in intracellular Ca2+.30 Ferroptosis is triggered by glutathione biosynthesis or glutathione peroxidase 4 (GPX4) activity inhibition28 31–33 (figure 1) and is associated morphologically with shrunken and electron-dense mitochondria.30 Three mechanisms in cancer cells have been reported to lead to ferroptosis: (1) depletion of glutathione and subsequent inactivation of GPX4, (2) Tf import or iron overload and (3) glutamine metabolism.30 31 34 Therefore, intracellular iron overload is clearly a major mechanism of Hb-induced and ferrous iron-induced neuronal ferroptosis after ICH, and decreasing iron accumulation in microglia might attenuate neuronal death. In a middle cerebral artery occlusion model of ischaemic stroke, ferroptosis contributes to neuronal death through tau–iron interaction.35 In addition to haemorrhagic and ischaemic stroke, ferroptosis has been shown to have significant implications in several other neurological diseases, such as Alzheimer’s disease, Parkinson’s disease and brain tumours.36
We have previously shown that ferroptosis contributes to neuronal death in the mouse collagenase-induced ICH model.10 Using transmission electron microscopy, we observed shrunken mitochondria in the brains of ICH mice, providing powerful evidence of ferroptosis. In OHSCs, we found that administration of ferrostatin-1 (Fer-1), a specific inhibitor of ferroptosis, prevented neuronal death and reduced iron deposition induced by Hb. In vivo, mice treated with Fer-1 after ICH showed marked brain protection and improved neurological function. We also found that Fer-1 reduced lipid ROS production and attenuated the increased expression level of prostaglandin-endoperoxide synthase 2 (PTGS2) and its gene product cyclooxygenase-2 (COX-2) in OHSCs and in vivo. Notably, COX-2 is highly expressed in neurons after ICH, and inhibition of COX-2 reduces ICH-induced secondary brain injury.37 38 Therefore, COX-2 might be a biomarker of ferroptosis. Based on these findings, we investigated whether a combination of cell death inhibitors could improve neuronal rescue. We found that Fer-1 in combination with other inhibitors that target different forms of cell death prevented Hb-induced cell death in OHSCs and human-induced pluripotent stem cell-derived neurons better than any inhibitor alone.10 Our most recent study showed that ferroptosis coexists with necrosis and autophagy in the mouse ICH brain.39 Therefore, it should be further determined whether a combination of inhibitors can improve ICH outcomes in animals better than one drug, and whether this strategy can be effective in clinical trials. In another new study, Zhang et al 40 showed that GPX4 expression was markedly reduced during the acute phase of ICH and that increasing GPX4 level was able to rescue neurons from ferroptotic death and improve outcomes in rats. These novel findings fill an important gap in knowledge regarding cell death after ICH and provide a vital foundation for cell death-based ICH treatment in the future.
Acknowledgments
The authors thank Claire Levine, MS, ELS, for assistance with manuscript preparation.
References
Footnotes
Contributors All authors wrote and revised the manuscript.
Funding This research was supported by the National Institutes of Health (R56 NS096549, R01 NS102583 and R21 NS102899 to J. Wang), the American Heart Association (Grant-in-Aid, 17GRNT33660766 to J. W ang and 18POST33970007 to J. Wan), and a Stimulating and Advancing ACCM Research grant from the Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.
Patient consent for publication Not required.