Article Text

Abstract
Stroke is the second and the leading most common cause of death in the world and China, respectively, but with few effective therapies. Nicotinamide phosphoribosyltransferase (NAMPT) is the rate-limiting enzyme for nicotinamide adenine dinucleotide (NAD) salvage synthesis in mammals, thereby influencing NAD-dependent enzymes and constituting a strong endogenous defence system against various stresses. Accumulating in-vitro and in-vivo studies have demonstrated the neuroprotective effect of NAMPT in stroke. Here, we review the direct evidence of NAMPT as a promising target against stroke from five potential therapeutic strategies, including NAMPT overexpression, recombinant NAMPT, NAMPT activators, NAMPT enzymatic product nicotinamide mononucleotide (NMN), and NMN precursors nicotinamide riboside and nicotinamide, and describe the relevant mechanisms and limitations, providing a promising choice for developing novel and effective therapeutic interventions against ischaemic and haemorrhagic stroke.
- neuroprotection
- nicotinamide adenine dinucleotide
- nicotinamide mononucleotide
- nicotinamide phosphoribosyltransferase
- stroke
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- neuroprotection
- nicotinamide adenine dinucleotide
- nicotinamide mononucleotide
- nicotinamide phosphoribosyltransferase
- stroke
Introduction
Stroke is the second most common cause of death1,2 and the third most common cause of disability-adjusted life-years (DALYs) worldwide,3 and the leading cause of death in China,4,5 with characteristics of high morbidity, high mortality, high disability rate, high recurrence rate and high medical expenses6 (box 1). Of all stroke case, approximately 78% are ischaemic stroke and others are haemorrhagic stroke.2 There is only one drug, tissue plasminogen activator (tPA), approved by Food and Drug Administration for the treatment of ischaemic stroke and no available drug for the treatment of haemorrhagic stroke. And, due to the narrow treatment window, contraindications and risk of complications, tPA is applicable for only about 3%–5% of stroke patients who meet the treatment criteria.2,7 Therefore, there is an urgent need for developing new and effective therapies for stroke patients. Stroke has a complex pathophysiology, which can cause a cascade of injury reactions with eventually cell death after the onset.2,8 At the same time, stroke can also mobilise the body’s defence mechanisms against impairments of stroke by improving cell survival, neurogenesis and functional recovery.2,9 Many previous studies have focused on the stroke injury mechanisms of excitotoxicity, oxidative and nitrosative stress, and inflammation to develop neuroprotective agents against stroke.2,10 However, these therapeutic strategies are still in the preclinical-to-clinical transition. Targeting the endogenous defence mechanisms against stroke has been received attention in the recent decade and considered as a novel target of stroke treatment.
The epidemiology of stroke
Stroke is an acute cerebrovascular disorder caused by sudden loss or deterioration of brain function due to the disruption of blood supply in the brain. Of all stroke case, approximately 78% are ischaemic stroke and others are haemorrhagic stroke.2 In ischaemic stroke, a clot occludes a brain vessel (most commonly the middle cerebral artery or its branches) with the cease of brain blood supply, causing a cascade of pathological events with energy failure, acidosis, excessive glutamate release, elevated intracellular Ca2+ level, generation of free radicals, blood–brain-barrier disruption, inflammation and eventually massive cell death.2,7,9 The haemorrhagic stroke has pathophysiological phases with arterial rupture, haematoma formation, haematoma enlargement and peri-haematoma oedema.8 Approximately 10% of ischaemic stroke victims and 38% of haemorrhagic stroke victims die within 30 days of stroke onset.7
Stroke is the second most common cause of death1,2 and the third most common cause of disability-adjusted life years (DALYs)3 worldwide. In the Global Burden of Diseases, Injuries, and Risk Factors Study (GBD, 2010), an estimated 16.9 million incident strokes occurred in 2010, which added to a pool of 33 million stroke worldwide.6,9 Although mortality rates and mortality-to-incidence ratios of stroke have decreased in the past two decades, the absolute number of people affected every year, stroke survivors, related deaths and DALYs lost are still increasing, particularly in low-income and middle-income countries.6 By 2030, there will be almost 12 million stroke deaths, 70 million stroke survivors, and more than 200 million DALYs lost globally6 with the continuing trend of stroke incidence, mortality and DALYs.6 In contrast to Western populations, stroke is the leading cause of death in China, where it accounted for 22.5% of deaths (about 2 million) in 2005.4 With the trends of increased stroke mortality in China, the death number will be doubled from 2 to 4 million in the next two decades.5 Having more than one-sixth of the world’s population, China has heavy personal, social and financial costs of stroke and related diseases in the context of having a high prevalence of stroke.
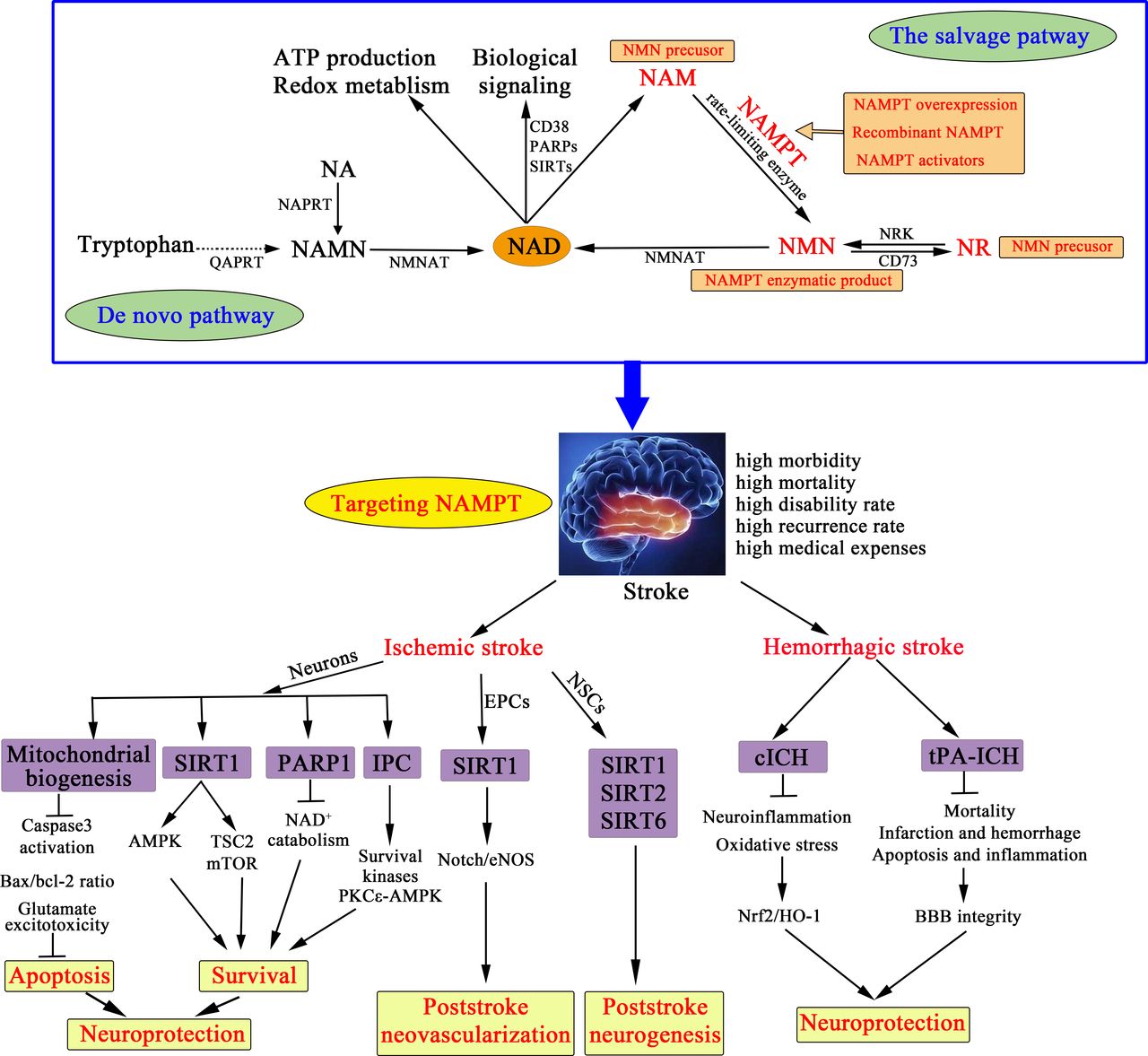
Nicotinamide phosphoribosyltransferase (NAMPT), also known as visfatin or pre-B cell colony-enhancing factor, is the rate-limiting enzyme for nicotinamide adenine dinucleotide (NAD) salvage synthesis in mammals (figure 1).2,11 The NAMPT–NAD axis plays a vital role in energy production, and connects to SIRTs (the most studied is SIRT1), poly-ADP-ribose polymerases (PARPs; the most studied is PARP1) and CD38 (a transmembrane enzyme), thereby constituting a strong endogenous defence system against various stresses.2,11 Experimental and clinical studies have demonstrated the change of NAMPT levels in stroke and other brain injuries. NAMPT is upregulated in the in-vitro cultured neurons of oxygen-glucose deprivation (OGD) model12 as well as in the in-vivo animal stroke models.12–14 Moreover, blood NAMPT levels in patients with ischaemic stroke,15–18haemorrhagic stroke19 20 19, 20 and traumatic brain injury21,22 are significantly increased, for instance the mean plasma concentration of NAMPT in ischaemic stroke patients is twofold to eightfold higher than that in persons without stroke.15–18

{kind=link}
Therapeutic strategies targeting NAMPT against stroke. There are two ways to maintain the cellular NAD level in mammals: the de novo pathway from tryptophan and the salvage pathway from NAM, NR and NA with different catalysing enzymes. NAMPT is the rate-limiting enzyme of NAD synthesis in the salvage pathway, thereby influencing NAD-dependent enzymes and regulating cellular metabolism, mitochondrial biogenesis and the adaptive response to inflammatory, oxidative, proteotoxic and genotoxic stresses. Accumulating in-vitro and in-vivo studies indicate that NAMPT is a therapeutic target against stroke from five potential therapeutic strategies: NAMPT overexpression, recombinant NAMPT, NAMPT activators, NAMPT enzymatic product NMN, NMN precursors NR and NAM. In ischaemic stroke, targeting NAMPT can confer neuroprotection via regulating mitochondrial biogenesis, activating SIRT1, inhibiting PARP1 activity and mimicking IPC. And, NAMPT related treatment exerts biological function in EPCs and NSCs to promote neovascularization and neurogenesis after ischaemic stroke. Remarkably, there is positive evidence of NAMPT in ICH, wherein NMN treatment can attenuate brain injury by activating Nrf2/HO-1 signalling pathway in the mouse model of cICH, and protect BBB integrity and attenuate brain infarction and haemorrhage in the mouse model of tPA-ICH, indicating NAMPT mediated neuroprotection in ICH. These preclinical evidences indicate targeting NAMPT can be a promising choice for developing novel and effective therapeutic interventions against stroke. BBB, blood–brain barrier; cICH, collagenase-induced intracerebral haemorrhage; EPCs, endothelial progenitor cells; IPC, ischaemic preconditioning; NA, nicotinic acid; NAD, nicotinamide adenine dinucleotide; NAM, nicotinamide; NAMN, nicotinic acid mononucleotide; NAMPT, nicotinamide phosphoribosyltransferase; NAPRT, nicotinic acid phosphoribosyltransferase; NMN, nicotinamide mononucleotide; NMNAT, nicotinamide mononucleotide adenylyltransferase; NR, nicotinamide riboside; NRK, nicotinamide ribose kinase; NSCs, neural stem cells; PARPs, poly-ADP-ribose polymerases; QAPRT, quinolinate phosphoribosyltransferase; SIRTs, sirtuins; tPA-ICH, tPA-induced cerebral infarction haemorrhagic transformation.
Amount of preclinical studies has demonstrated targeting NAMPT can strongly suppress ischaemia-neuronal injury to exert neuroprotection in the acute phase of ischaemic stroke, and promote neovascularisation and neurorestoration in the sub-acute and chronic phase of ischaemic stroke (figure 1). Moreover, in the two models of intracerebral haemorrhage (ICH), collagenase-induced ICH (cICH) and tPA-induced haemorrhagic transformation after ischaemic stroke (tPA-ICH), NAMPT enzymatic product nicotinamide mononucleotide (NMN) exerts neuroprotection (figure 1). In this review, we summarise direct evidence of NAMPT as a target against stroke from five therapeutic strategies, including NAMPT overexpression, recombinant NAMPT, NAMPT activators, NAMPT enzymatic product NMN, and NMN precursors nicotinamide riboside (NR) and nicotinamide (NAM), and describe the relevant mechanisms, demonstrating the therapeutic value of NAMPT-related agents in the treatment of stroke (figure 1). In addition, we discuss the limitations of NAMPT in stroke, hoping to promote the preclinical-to-clinical translation of NAMPT-related agents in ischaemic and haemorrhagic stroke.
NAMPT overexpression
Our group previously gave the first demonstration for preferential expression of NAMPT in neurons as opposed to astrocytes, and found the significant upregulation in the peri-infarct area and infarct core of middle cerebral artery occlusion (MCAO) models.12 NAMPT inhibition by a highly-specific NAMPT inhibitor, FK866, aggravates brain infarction in experimentally cerebral ischaemia rats, whereas NAMPT overexpression (approximately 2.0-fold) by injection of lentiviral vectors encoding NAMPT (LV-NAMPT) in the rat cortex and hippocampus significantly decreases infarct size and brain cell death, and ameliorates neurological deficit score.12 Further, AMPKα2 and SIRT1 knockout mice are used to investigate the underlying mechanisms of NAMPT neuroprotection.12 We found NAMPT overexpression and knockdown regulate neuronal survival via the AMPK pathway, due to the neuroprotection of NAMPT is abolished in AMPKα2−/- neurons.12 And, SIRT1 is essential for NAMPT-induced AMPK activation and neuroprotection, because AMPK activation by NAMPT overexpress disappears in SIRT1−/− neurons.12 Moreover, NAMPT overexpression-induced neuroprotection is abolished in SIRT1+/− and AMPKα2-/- mice. Thus, NAMPT overexpression reduces OGD-induced neuronal injury via SIRT1-dependent AMPK pathway.12
Considering genetic deletion of NAMPT is lethal in mice,23 NAMPT knockout heterozygous mice (Nampt+/− mice),24,25 transgenic mice overexpressing NAMPT (Nampt-Tg mice)26,27 and H247A-mutant dominant-negative NAMPT (ΔNampt-Tg mice)26,27 are generated to explore the role of NAMPT in stroke. In the photothrombosis-induced ischaemia model, knockout of NAMPT in Nampt+/− mice exacerbates ischaemic brain injury with lower NAMPT expression and NAD+ level, and higher density of degenerating neurons in the penumbra than that in wild-type (WT) mice,24 wherein NAMPT exerts neuroprotective effect in ischaemia through its enzymatic activity for NAD+ production that can ameliorate mitochondrial dysfunction.25 Our group have generated Nampt-Tg mice and ΔNampt-Tg mice, and found that Nampt-Tg mice, but not ΔNampt-Tg mice, exhibit enhanced capillary density, increased number of proliferating endothelial cells, improved blood flow recovery, augmented collateral arterioles in the ischaemic limb and demonstrated intracellular NAMPT–NAD+/−–SIRT1 cascade improves postischaemic vascular repair by modulating Notch signalling in endothelial progenitors.26 New-formed blood vessels in the brain penumbra of Nampt-Tg mice are more than that in WT mice at 14 days after MCAO,28 demonstrating improved poststroke angiogenesis. Furthermore, compared with WT mice, NAMPT-Tg mice show enhanced number of neural stem cells, improved neural functional recovery, increased survival rate and accelerated body weight gain after MCAO, which are not observed in ΔNampt-Tg mice.27 Furthermore, neuron-specific transgenic mice overexpressing NAMPT significantly reduce infarct volume by 65% and improve long-term neurological outcomes compared with WT littermates in the MCAO model.14 And neuronal overexpression of NAMPT increases the area of myelinated fibres in the striatum and corpus callosum, indicating that NAMPT protects against white matter injury.14 In the glutamate-induced excitotoxicity model of cultured neurons, overexpression of NAMPT significantly promotes neuronal survival, reduces the translocation of apoptosis-inducing factor from mitochondria to nuclei and inhibits caspase-3 activation through inhibiting caspase-dependent and independent apoptotic signalling pathways.29 And also, overexpression of NAMPT can suppress glutamate-induced mitochondrial fragmentation, the loss of mitochondrial DNA content and the reduction of PGC-1 and NRF-1 expressions by suppressing mitochondrial damage and dysfunction.29
Besides, ischaemic preconditioning can also stimulate NAMPT and elevate NAD level in purified mitochondrial fractions via a PKC-ε-dependent manner,30 suggesting that the mitochondrial NAMPT regulated NAD pool is involved in the neuroprotection of the ischaemic conditioning.30
Recombinant NAMPT
Considering that NAMPT can cross the blood–brain barrier (BBB)14 and the BBB is relatively open at an early stage of postischaemic stroke in both animals and patients,31,32 recombinant NAMPT proteins represent a clinically feasible tool that can be generated and delivered under controlled settings in stroke. Our group first demonstrated extracellular recombinant mouse WT NAMPT, but not mouse H247A-mutant enzymatic-dead NAMPT, has the enzymatic activity in vitro, and significantly attenuates the detrimental effect of OGD on the cell viability and apoptosis in both cultured mouse neuron and glia, wherein treatment of neutralising antibody abolishes the protective effect on cell viability.13 Moreover, in the transient global cerebral ischaemia model, administration of NAMPT at the time of reperfusion can reduce neuronal necrosis and apoptosis of hippocampus CA3 cells,33 protect against ischaemia/reperfusion injury via modulation of Bax/bcl-2 ratio and prevention of caspase-3 activation by intracerebroventricular (ICV) injection at hippocampal CA3 area,34 and reduce hippocampal CA1 cells death and improves learning and memory deficits by ICV injection at hippocampal CA1 area.35
NAMPT activators
Simultaneously with the identification of NAMPT as a therapeutic target against stroke, we created an in-vitro high-throughput screening assay to discover novel NAMPT-active compounds.36 We have screened approximately 55 000 small-molecule chemicals with obtaining 348 compounds with NAMPT activity <40% and 495 compounds with NAMPT activity >125%, and verified a potent inhibitor MS0 and a fluorescent compound M049-0244.36–38 With crystal structure-based comparison of two NAMPT inhibitors MS0 and FK866 in their biological activity and molecular binding mode,39 the deep digging of mode action of NAMPT inhibitors as well as fluorescent compound helps to understand the action mode of NAMPT and image NAMPT, providing possibility for developing new drugs of NAMPT and detecting NAMPT distribution.2,36–38
Moreover, we also investigated the effect of a potential NAMPT stimulator P7C3-A20 on ischaemic stroke.40 P7C3-A20, an aminopropyl carbazole derivative, has been reported to have proneurogenic and neuroprotective effects in various brain disease models.11 In the successive studies, P7C3-A20 is found to protect hippocampal mature neurons and substantia nigra dopaminergic neurons from cell death in the Parkinson’s Disease model, protect ventral horn spinal cord motor neurons from cell death in the Amyotrophic Lateral Sclerosis model, augment hippocampal neurogenesis following focal ablation of hippocampal stem cells in the depression-prone ghrelin receptor-null mouse model, and increase proliferating neurons and improve cognitive function after traumatic brain injury.11 Combined with these studies and the enhanced flux of NAD by P7C3 compounds in mammalian cells, P7C3 family is proposed as a therapeutic approach to treat cerebral ischaemia. As such, our group first demonstrated P7C3-A20 increases neuronal viability in the in-vitro OGD model, and reduces cerebral infarction and restores the brain NAD level in the in-vivo MCAO model of acute ischaemic stroke.40 Further, the effectiveness of P7C3-A20 treatment on chronic histopathological and behavioural outcomes and neurogenesis after ischaemic stroke has been also explored.41,42 P7C3-A20 treatment for 7 days immediately after MCAO improves rat performance in sensorimotor cylinder and grid-walk tasks, and in a chronic test of spatial learning and memory after stroke. P7C3-A20 treatment also significantly decreases cortical and hippocampal atrophy, increases neurogenesis in the SVZ and hippocampal DG, and restores NAD to sham level after ischaemic stroke.41 Moreover, delayed P7C3-A20 treatment, at 6-hour post-reperfusion of MCAO, significantly improves stroke-induced sensorimotor deficits in motor coordination and symmetry, as well as cognitive deficits in hippocampus-dependent spatial learning, memory retention, and working memory. In the cerebral cortex, delayed P7C3-A20 treatment significantly increases tissue sparing at 7 weeks after stroke and reduces hemispheric infarct volumes at 48 hours post-reperfusion.42 Importantly, P7C3-A20 treatment in adult male rhesus monkeys for 38 weeks provides sustained plasma exposure, well tolerates and safely elevates BrdU+ survival neurons in the hippocampus of non-human primates, suggesting that the neuroprotective efficacy of P7C3 compounds is likely to translate to humans as well.43 Besides, in a paclitaxel-induced peripheral neuropathy model, P7C3-A20 abrogates neuropathic pain, protects peripheral nociceptive neurons from damage, improves general health, reduces attrition associated with PTX treatment and enhances NAMPT-mediated NAD recovery in response to cellular damage.44 And those beneficial effects can be abolished by the NAMPT inhibitor, suggesting the augmentation of NAMPT activity is required for P7C3-A20-mediated neuroprotection.44 Taken together, P7C3-A20 may be new therapeutic approach for stroke via the augmentation of NAD level and NAMPT activity, which safely augments NAD level in vivo and protect from neuronal injury in the acute and chronic phase of stroke.
Nicotinamide mononucleotide
As the direct enzymatic product of NAMPT, NMN has the property of small-molecule that can be easy to administer to modulate brain NAD level in the MCAO model.2,12,27 ICV injection of NMN attenuates cerebral infarction, and prevents neurotoxic effects of NAMPT inhibitor FK866 in the rat MCAO model.12 In the in-vitro ischaemic model of OGD-treated neurons, NMN can protect neurons against injury with decreased proapoptotic protein levels and increased antiapoptotic protein levels.12 Moreover, early NMN administration with the first dose at 30 min post MCAO for 7 days decreases brain infarction and neurological deficit, enhances animal survival and accelerates body weight recovery.27 Although delayed administration of NMN for 7 days with the first dose at 12 hours post MCAO is unable to decrease brain infarction and neurological deficit and affect the body weight, delayed NMN treatment reduces MCAO-induced death during the first week post MCAO, improves neuronal recovery and increases neurogenesis in the subventricular zone and dentate gyrus of MCAO mice.27 Besides, NMN induces proliferation and pro-differentiation of neural stem cells in the in-vitro cultured neurosphere.27 A study by another group also demonstrated the neuroprotection of NMN. In the mouse model of transient forebrain ischaemia, NMN can ameliorate the hippocampal CA1 injury, improve neurological outcome and prevent the increase of PAR formation and NAD+ catabolism.45 Besides, a recent study found deletion NAMPT of projection neuron leads to motor dysfunction and death of mice. And NAMPT deletion impairs mitochondrial function and synaptic transmission of neuromuscular junctions. When treated with NMN, conditional knockout mice of NAMPT exhibit reduced motor function deficits and prolonged lifespan, suggesting neuronal NAMPT plays an essential role in mitochondrial bioenergetics, motor function and survival.46
Based on the above results of NMN in the ischaemic stroke, we further explored the effects of NMN in the two models of ICH (including cICH and tPA-ICH). In the mouse model of cICH, although administration of NMN at 30 min post cICH shows no effect on haematoma volume and haemoglobin content, NMN treatment significantly reduces brain oedema, cell death, oxidative stress and neuroinflammation in brain haemorrhagic area by activating Nrf2/HO-1 signalling pathway.47 And, a prolonged NMN treatment for 7 days can markedly promote the recovery of body weight and neurological function.47 Considering tPA is the only approved pharmacological therapy for acute ischaemic stroke but followed with a major limitation of haemorrhagic transformation, our group established tPA-ICH model to explore the effect of NMN.48 In the mice infused with tPA at 5 hours post-ischaemia, there is significant increase in mortality, brain infarction, brain oedema, brain haemoglobin level, neural apoptosis and neuroinflammation, all of which can be significantly prevented by NMN administration.48 Mechanistically, NMN can maintain BBB integrity, whereas delayed tPA treatment increases BBB permeability.48
Taken together, as an endogenous substance with relatively high safety, NMN can be a promising drug candidate for the treatment of ischaemic and haemorrhagic stroke.
NR and NAM
NR and NAM are the NMN precursors in the NAD salvage synthesis pathway, both of which can replenish and elevate NAD level.2 Intracortical administration of NR shows better neuroprotective effect than NAD+ in excitotoxicity-induced axonal degeneration.49 Our group has demonstrated NR treatment can significantly reduce brain infarction volume and protect against brain injury in the rat MCAO model of ischaemic stroke (China patent: ZL201510406371.4). And, NR also exerts benefits in the model of Parkinson’s50 and Alzheimer’s Disease.51
In the rat model of permanent MCAO, NAM reduces neuronal infarction in a dose-specific manner and reduces infarcts when administered up to 2 hours after the onset of permanent occlusion.52 And, NAM can delay the onset of stroke and reduce infarction volume when administrated to stroke-prone spontaneously hypertensive rats before the ischaemic event.53 NAM pretreatment plays antioxidative action and alleviates mitochondrial membrane potential in the infarct penumbra induced by MCAO.53 It is important to realise that NAM shows opposite effects with different dosage. In one dosage-reaction study of rat temporary MCAO model, NAM treatment at 20 mg/kg within 0–6 hour after reperfusion shows potent neuroprotective effects, but administration of NAM 60 mg/kg at the onset of reperfusion drastically accelerates brain damage.54 The diverse functions with NAM treatment may be due to the role of NAM in different signalling, wherein NAM not only inhibit PARP,55 but also inhibit the activity of the SIRT2 family of histone/protein deacetylases.56 Recently, it is reported that NAM administration significantly reduces infarct area and enhances fractional anisotropy in the ipsilesional internal capsule induction than the other groups at 7 and 14 days after stroke, respectively.57 NAM also promotes the expression of NAD+, BDNF and remyelination markers and improve functional recovery, which can be abolished by the administration of NAMPT inhibitor FK866 or TrκB antagonist ANA-12.57 Therefore, NAM administration improves remyelination after stroke via the NAD+/BDNF/TrκB pathway.
Limitations of NAMPT in stroke
Above evidence demonstrated NAMPT is a promising target for developing therapeutic interventions against ischaemic stroke and haemorrhagic stroke. Although the safety of P7C3-A20 has been studied in adult male rhesus monkeys, the efficacy of above NAMPT-related agents is still in the preclinical study phase, primarily studied in the in-vivo MCAO model of rodent animals and in-vitro OGD model of primary cortical neurons. Recent breakthroughs in human pluripotent stem cell-derived humanised neurons and cerebral organoids not only show potential in the replacement for expensive non-human primate animals, but also provide a valuable platform for investigating the neuroprotection of NAMPT in humanised stroke model. Further studies need to answer the efficacy of NAMPT-related agents in the humanised neurons/cerebral organoids, non-human primates and ultimately humans. Besides, the role of NAMPT in stroke is only explored as monotherapy. It is still unclear whether NAMPT-based combination therapy with other neuroprotective agent or anti-inflammatory treatment is more effective to cure stroke.
Conclusion
Accumulating in-vitro and in-vivo studies indicate that NAMPT is a therapeutic target in ischaemic stroke. NAMPT–NAD signalling represents a strong endogenous defence system against ischaemia-induced energy deletion, neural cell death, and eventually brain infarction. Our group also provides first-hand evidence of NAMPT in ICH, wherein NMN attenuates brain injury after ICH by activating Nrf2/HO-1 signalling pathway, and protects BBB integrity and attenuates delayed tPA-induced haemorrhagic transformation after cerebral ischaemia. From the elaboration of NAMPT overexpression, recombinant NAMPT, NAMPT activators, NAMPT enzymatic product NMN, NMN precursors NR and NAM in stroke, targeting NAMPT can be a promising choice for developing novel and effective therapeutic interventions against stroke. For further preclinical-to-clinical translation, future work is needed to answer more scientific questions, especially the effect of NAMPT-related agents in the humanised neurons/brains or non-human primates, providing substantial preclinical evidence to promote new drug discovery of stroke treatment.
References
Footnotes
Contributors C-YM formulated the concept and reviewed the manuscript, S-NW and C-YM drafted the manuscript.
Funding This work was supported by grants from the National Natural Science Foundation of China for Distinguished Young Scholars (30525045), National Natural Science Foundation of China Major Project (81730098), National Natural Science Foundation of China (81373414), National Basic Research Program of China (2009CB521902), National Science and Technology Major Project (2009ZX09303-002), Medical Innovation Major Project (16CXZ009) and Shanghai Science and Technology Commission Project (16431901400).
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement No additional data are available.
Patient consent for publication Not required.