Article Text

Abstract
Objective Subarachnoid haemorrhage (SAH) accounts for 3% of all strokes, and is associated with significant morbidity and mortality. There is growing evidence implicating apolipoprotein E (apoE) in mediating adaptive anti-inflammatory and neuroprotective responses following ischaemic and traumatic brain injury. In the current study, we test the efficacy of a small apoE mimetic peptide, CN-105 in a murine model of SAH.
Methods Mice subjected to SAH received repeated intravenous injections of CN-105 every 12 hours for 3 days, with the first dose given 2 hours after injury. Daily functional outcomes were assessed by rotarod and neurological severity score. Haemorrhage grade and cerebral vascular diameters were measured at 5 days post-SAH. Cerebral microgliosis, neuronal degeneration and survival were analysed at 5 and 35 days post-SAH, respectively.
Results CN-105 reduces histological evidence of inflammation, reduces vasospasm and neuronal injury and is associated with improved long-term behavioural outcomes in a murine model of SAH.
Conclusions Given its favourable pharmacokinetic profile, central nervous system penetration and demonstration of clinical safety, CN-105 represents an attractive therapeutic candidate for treatment of brain injury associated with SAH.
- apolipoprotein E
- subarachnoid hemorrhage
- vasospasm
- microgliosis
- inflammation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Although subarachnoid haemorrhage (SAH) represents a minority of strokes, it is a devastating disease associated with high rates of mortality (40% at 1 month) and morbidity (only 50% of survivors return to work).1 SAH also tends to occur at a younger age than ischaemic stroke2 and is thus associated with high personal and societal costs. Ultimately, functional outcomes following aneurysmal SAH are influenced by many factors, including the development of delayed cerebral ischaemia (DCI).2 3 Although the exact pathophysiology of DCI remains incompletely understood, DCI is associated with changes in the cerebral vasculature, including a proliferative angiopathy associated with smooth muscle and endothelial proliferation, vasospasm and microthrombosis. Vasospasm-related delayed ischaemic neurological deficits have been estimated to account for 40% of deaths in people surviving initial aneurysm rupture and are strongly associated with unfavourable outcomes.4 It has been suggested that mechanisms that activate within minutes after SAH lead to early secondary brain injury and may play an important role in the pathogenesis of delayed ischaemic injury and poor outcome.5
Unfortunately, although great advances have been made with regard to early endovascular ablation of cerebral aneurysms, the medical treatment of cerebral vasospasm and management of secondary brain injury remains largely supportive. In particular, there are no pharmacological interventions that have been demonstrated to reduce the incidence of delayed ischaemic deficit due to vasospasm. Treatment with nimodipine, an L-type calcium channel blocker, has been associated with incremental improvement in functional outcomes and is widely regarded as standard of care, although it has not been demonstrated to reduce angiographic vasospasm.6 7 Similarly, recent clinical trials evaluating pharmacological therapies designed to reduce vascular changes associated with SAH have failed to improve outcomes.8–11
One innovative approach to improve outcomes following aneurysmal SAH is administration of apolipoprotein E-mimetic drugs. Apolipoprotein E (apoE) is the primary apolipoprotein produced within the brain, where its secretion is upregulated after injury. ApoE has three common human isoforms (apoE2, apoE3 and apoE4) which differ by single cysteine to arginine interchanges at residues 112 and 15812. In addition to its role in cholesterol transport, several studies have suggested that apoE may exert isoform-specific modulations of the inflammatory response of the injured central nervous system (CNS). In particular, presence the APOE4 allele has been associated with poor functional outcome in a number of acute brain injuries, including SAH.13–16 One plausible explanation for these isoform-specific effects is that apoE modulates glial activation and neuroinflammatory cascades in an isoform-specific fashion. Indeed, the apoE4 isoform is associated with reduced neuroprotective and immunomodulatory properties as compared with apoE3.16–21
Thus, given the role of inflammation and secondary brain injury in SAH, one therapeutic strategy is to harness the beneficial effects of endogenous apoE. Unfortunately, due to its size, systemically administered apoE does not readily cross the blood brain barrier. To address this problem, we created small apoE peptides derived from the receptor binding region that maintained the neuroprotective and anti-inflammatory effects of the intact protein.22 23 In fact, as proof of principle, we have demonstrated that these apoE peptides reduce histological evidence of vasospasm and improve functional outcome in a preclinical models of SAH.24–26
Recently, we have created CN-105, a 5 amino acid peptide derived from the receptor binding face of the apoE receptor binding region. This second generation apoE mimetic is associated with increased potency and CNS penetration27 and improves outcome in several acute CNS pathologies that contribute to secondary cerebral injury following SAH, including ischaemia,28 mechanical brain injury29 and intraparenchymal haemorrhage.27 Moreover, CN-105 has recently been translated to phase I clinical trials, where it was demonstrated to be safe, well tolerated and possessed favourable pharmacokinetics.30 We now test the hypothesis that systemic administration of CN-105 will reduce the vasculopathy and functional deficits associated with a validated preclinical model of aneurysmal SAH.
Materials and methods
All animal procedures were approved by the Duke University Institutional Animal Care and Use Committee with established guidelines. Ten to 12-week-old C57BL/6J male mice (Jackson Laboratories, Bar Harbor, Maine, USA) were randomised into experimental groups. Body weight was recorded daily. All procedures and assessments were performed in a blinded fashion with regard to treatment assignment.
Peptide synthesis and administration
CN-105 (Ac-VSRRR-amide) was synthesised by Polypeptide (San Diego, California, USA) to a purity of >99% as previously described and was dissolved in sterile saline immediately before use. Based on efficacy in prior studies,27–29 0.05 mg/kg of CN-105 dissolved in 100 µL volume of saline was administered via tail vein injection every 12 hours for 3 days with the first dose given 2 hours following SAH induction.
Mouse model of SAH
Our murine injury model was adapted from a previously described model of SAH in mouse.31 Ten to 12-week-old male mice underwent anaesthesia induction in a chamber with 4.6% isoflurane. The trachea was intubated and the lungs were mechanically ventilated with 1.6% isoflurane in 30%/70% (O2/N2). Rectal temperature was continuously monitored and surface heating or cooling was servoregulated to maintain a core temperature of 37.0°C±0.2°C during the entire surgical procedure. A midline incision was made in the neck, and the right common carotid artery was exposed. The right external carotid artery (RECA) was isolated and ligated, leaving a small stump attached to the common carotid artery. A blunted 5–0 monofilament nylon suture, 15 mm in length was introduced into the RECA stump and advanced into the right internal carotid artery to the bifurcation of the right anterior cerebral artery (RACA) and right middle cerebral artery (RMCA). Here, resistance was encountered and the monofilament was advanced 3 mm further to perforate the RACA, resulting in subarachnoid bleeding. After the removal of the monofilament, the RECA stump was tied close and the skin was closed with sutures. Animals were allowed to recover spontaneous ventilation with subsequent extubation. Following recovery in a warm non-stimulating environment, the mice were allowed free access to food and water.
Neurological evaluation following SAH
Behavioural outcomes were assessed as previously described.32 Vestibulomotor function was assessed using an automated rotarod (RR) (Ugo Basile, Comerio, Italy). On the day prior to SAH induction, baseline RR performance was obtained for all mice. The animals received training by placing the mice on the RR apparatus for a single 300 s training period with the RR set in an accelerating rotational speed mode (4–40 rpm). After training, RR latencies (the time the animal remains on the accelerating rotating bar) are recorded for three trials per day. Following training, the average time to fall from the rotating cylinder over three trials was recorded as baseline latency. On days 1 through 5 after injury, the mice were given three trials daily with accelerating rotational speed (intertrial interval >15 min). The average latency to fall from the rod or to continuously rotate twice holding on to the rod was recorded for each animal each day.
A second outcome assessed was the neurological severity score. The score was based on an assessment of motor components derived from spontaneous activity, symmetry of limb movements, climbing and balance and coordination, with each component being scored from 0 to 3. Sensory components were used to analyse body proprioception and tactile and vibrissa responses to stimuli. These components were scored from 1 to 3. On the day prior to SAH, baseline neurological severity score was obtained for all mice. Assignment of the neurological severity score evaluation was repeated daily for 5 days postoperatively.
Haemorrhage grade
Brains were analysed by a blinded observer under light microscopy to determine SAH severity as a function of haemorrhage size and clot density. Haemorrhage area was scored as: (1) SAH extends anteriorly <1.0 mm from MCA–ACA bifurcation; (2) SAH extends >1.0 mm anteriorly from bifurcation; (3) SAH extends >1.0 mm anteriorly from bifurcation. Haemorrhage density was scored as 1 (underlying brain parenchyma visualised through clot) or 2 (underlying brain parenchyma not visualised through clot). Haemorrhage grade (2–5) was determined by the sum of the size and density scores as previously described.31
Cerebral vascular diameter measurement after SAH
After the completion of behavioural examination on post-SAH day 5, cerebral vascular perfusion was performed as described previously.31 Mice were placed in a chamber and induced with 4.6% isoflurane as above. The chest was opened to allow cannulation of the proximal aorta. Silicon tubing (0.76 mm internal diameter) was used to deliver infusion solutions at physiological pressures in a pulsatile manner. Typical infusion solutions consisted of 30 mL of saline, 30 mL of 4% formalin and 10 mL of ink/gelatin mixture solution, which were infused in that order. The mouse was refrigerated for 24 hours to allow solidification of the gelatin within the vasculature. Then, the mouse brain was harvested and stored in 10% formalin. The cerebral vasculature was imaged using a video-linked dissecting microscope controlled by an image analyzer (MCID Elite; Interfocus Imaging, Linton, UK). The lumen diameter of the RMCA, left middle cerebral artery (LMCA) at 1 mm proximal to the MCA–ACA bifurcation and the lumen diameter of basilar artery (BA) at 1 mm proximal to the rostral cerebellar artery were measured.
Immunohistochemistry and stereological analysis
Histological procedures were slightly modified from previously described.31 Mice were anaesthetised with isoflurane and perfused intracardially with saline at 5 days post-SAH for Fluoro-Jade B (FJB) (Histo-Chem, Jefferson, Arkansas, USA) and F4/80 (rat monoclonal, 1:10,000; Serotec, Raleigh, North Carolina, USA) staining, or 35 days post-SAH for staining of antineuronal-specific nuclear protein antibody (NeuN) (mouse monoclonal, 1:30 000; Chemicon, Temecula, California, USA). Brains were removed and immersion fixed in 4% buffered formaldehyde for 24 hours then transferred into 1× PBS and stored at 4°C. After brains were placed in 30% sucrose buffer for at least 24 hours, frozen coronal sections (30 µm) were cut and collected using a sliding microtome and stored in cryoprotectant solution containing ethylene glycol, sucrose and sodium phosphate. Equally spaced sections 240 µm apart were used for immunohistochemistry.
For F4/80 and NeuN staining, free-floating sections were incubated in 1% hydrogen peroxide, permeabilised by 0.1% saponin and blocked with 10% goat serum. Primary antibody was applied overnight at 4°C and then incubated in secondary antibody (biotinylated IgG antibody, 1:3000; Vector Laboratories, Burlingame, California, USA) for 2 hours, followed by avidin-biotin-peroxidase complex treatment for 1 hour (ABC kit; Vector Laboratories,). Staining was visualised with diaminobenzidine (DAB; Vector Laboratories). Between treatments, tissue was washed 3 times for 5 min each with Tris-buffered saline. The sections were then mounted onto charged slides and counterstained with haematoxylin (Fisher Scientific, Fair Lawn, New Jersey, USA).
For FJB (Histo-Chem) staining, sections are mounted onto gelatin charged slides and dehydrated, stained with 0.0001% FJB in 0.1% acetic acid (Schmued and Hopkins, 2000) and cover-slipped using DPX mounting media (Fluka, Milwaukee, Wisconsin, USA). Slides containing hippocampus were examined for degenerating neurons using an epifluorescent microscope (Nikon, Tokyo, Japan) with a medium band blue excitation (Nikon B-2A, 450–490 nm) filter set. For FJB positive cell quantification, degenerating neurons were quantified at 20×magnification by randomly counting the area of 7258 µm2 of the pyramidal layer of hippocampus CA1+CA2 of FJB-positive neurons in every eighth section of brain hippocampus by an observer blinded to group assignment.
Quantification of hippocampal F4/80 and NeuN positive cells was conducted using a Nikon 218912 light microscope interfaced with the StereoInvestigator software package (MicroBrightField, Williston, Vermont, USA) as described previously.29 The number of stained cells per mm3 was estimated by the optical fractionator method, which is an unbiased counting method and independent of the size, shape and orientation of the cells to be counted. The parameters of the fractionator-sampling scheme were established in a pilot experiment and were uniformly applied to all animals. Before counting, all the slides were coded to avoid experimenter bias. As determined by StereoInvestigator, we chose four coronal sections (30 µm thick) spaced eight sections apart along the dorsal hippocampal formation by systematic random sampling. This number of sections proved sufficient to provide a coefficient of error between 0.09 and 0.11. The total number of F4/80 or NeuN immunopositive cells was calculated per hippocampal volume of 960 µm thickness.
Statistical analysis
RR latencies and neurological severity scores were compared using repeated-measures analysis of variance with time as the repeated variable. Luminal diameters were compared by analysis of variance. When indicated by significant F ratio, pairwise comparisons were made and correction for repeated measures was made with the posthoc Tukey test. The numbers of FJB, F4/80 and NeuN cell counts were compared between groups by the Independent Samples Test. Parametric values were expressed as mean±SEM. Significance was assumed when p<0.05.
Results
SAH bleeding and severity is consistent in the model
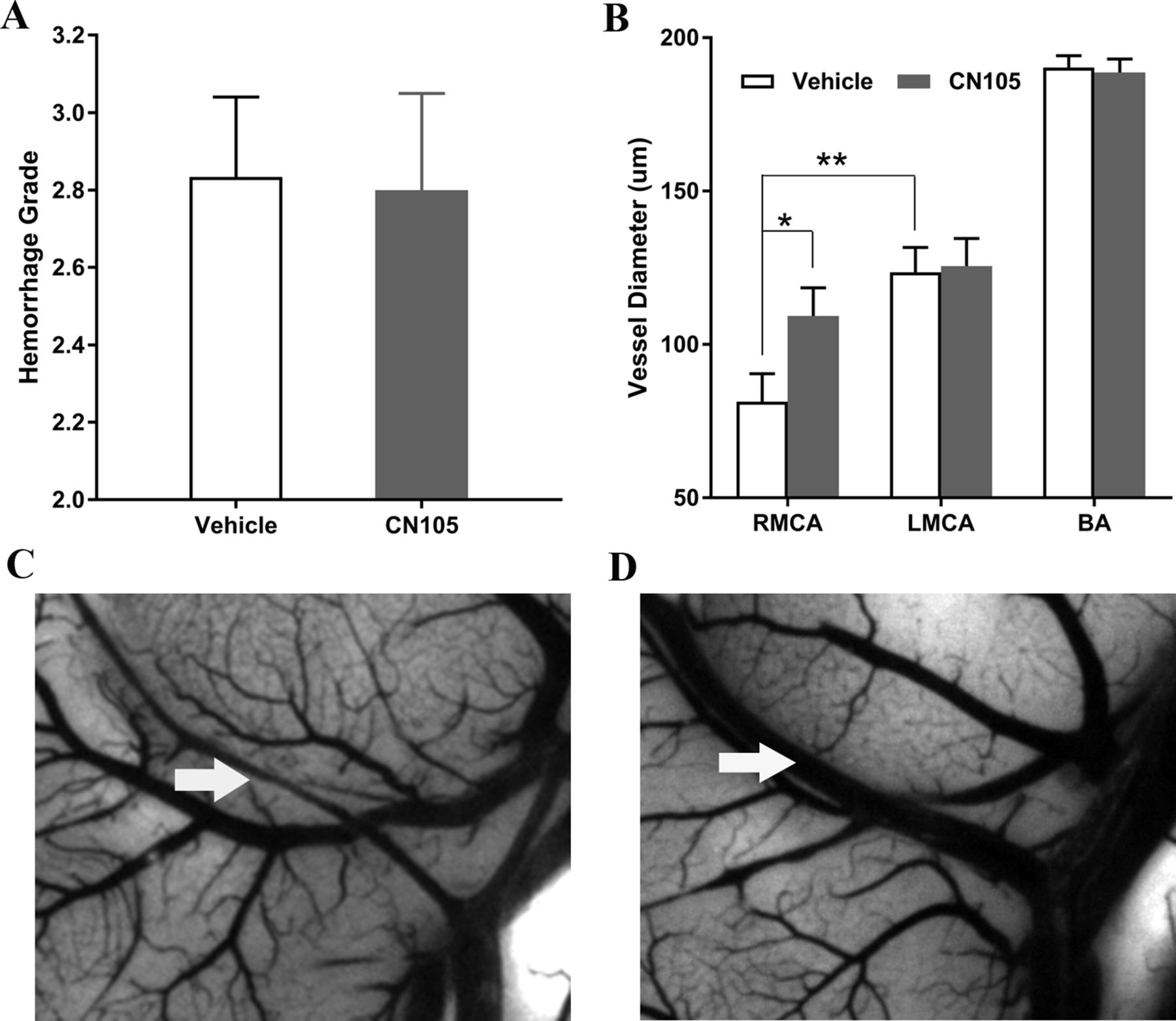
The mean SAH size and severity scores were similar in the control animals across experiments and between CN-105-treated and untreated animals. As demonstrated in figure 1A, the mean SAH size was 2.80±0.249 and 2.83±0.207 and the severity scores were 2.83±0.207 and 2.80±0.249 for the CN-105-treated and vehicle groups, respectively. There was no significant difference in these measures (p=0.918 bleed size, p=0.918 for severity score), demonstrating consistency of the size and severity of SAH across treatment groups.

Haemorrhage volume was similar between CN-105-treated and saline-treated mice (A); SAH successfully induced vasospasm (B. RMCA 81.39±31.52 µm vs LMCA 124.46±27.53 µm; p=0.002) in vehicle-treated mice and CN-105 significantly reduced RMCA vasospasm at 5 days after SAH compared with Vehicle group (vehicle 81.39±31.52 µm; VR55 109.30±28.95 µm; p=0.044); there was no difference between the groups in LMCA and BA diameter used as a control as these vessels were remote from the haemorrhage. Ink/gelatin casting performed at 5 days after SAH (C,D). BA, basilar artery; LMCA, left middle cerebral artery; RMCA, right middle cerebral artery; SAH, subarachnoid haemorrhage.
CN-105 reduces vasospasm after SAH
We next assessed whether treatment with CN-105 was associated with a reduction in vasospasm, as assessed by comparisons of RMCA luminal diameter, using the specific methodology that we have previously defined.31 32 Five days after induction of SAH, 22 animals (CN-105 n=10; vehicle n=12) were perfused with ink/gelatin and the brains were harvested for vascular luminal diameter assessment. In vehicle-treated animals, induction of SAH was associated with a reduction in luminal diameter of the RMCA, as compared with LMCA (RMCA 94.08±32.90 µm; LMCA 124.46±27.53 µm; p=0.002). Animals treated with CN-105 demonstrated significantly reduced ipsilateral RMCA luminal narrowing as compared with control-treated animals (vehicle 81.39±31.52 µm; CN-105 109.30±28.95 µm; p=0.04). There was no difference between the two treatment groups in LMCA diameter (vehicle 123.58±27.96 µm; CN-105 125.5±28.5 µm; p=0.87) or BA diameter (vehicle 190.29±13.35 µm; CN-105 188.7±13.9 µm; p=0.78), which were remote from the subarachnoid clot (figure 1B–D). In summary, these results demonstrate the consistent development of vasospasm in blood vessels adjacent to subarachnoid clot which was attenuated by CN-105 treatment.
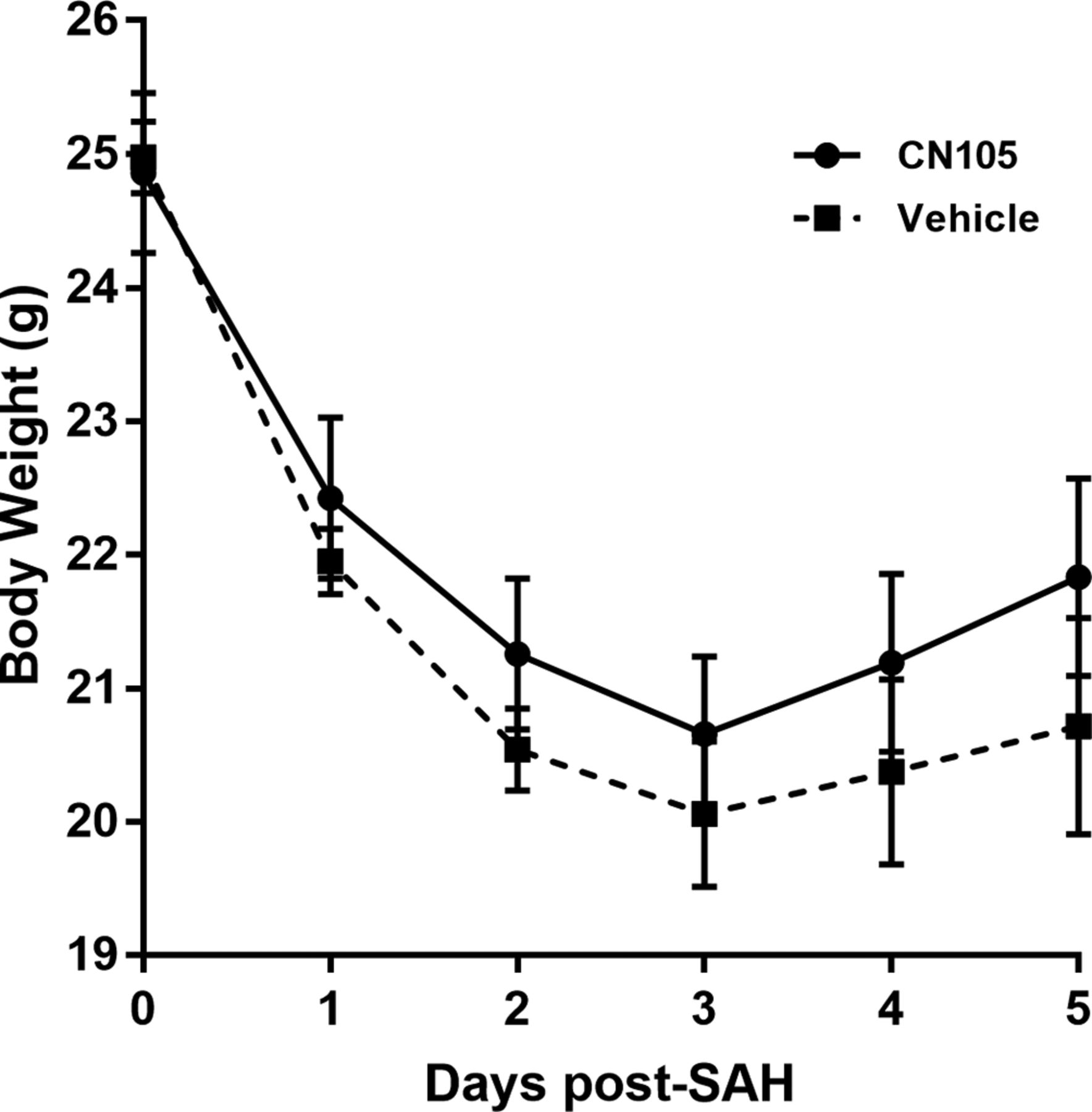
CN-105 is associated with earlier body weight recovery
After SAH, the mice generally lost an average of 3.6±0.46 g of body weight. Administration of CN-105 was well tolerated and was associated with better body weight recovery than vehicle-treated mice over the first 5 days after SAH (figure 2, n=12/group; p=0.05).
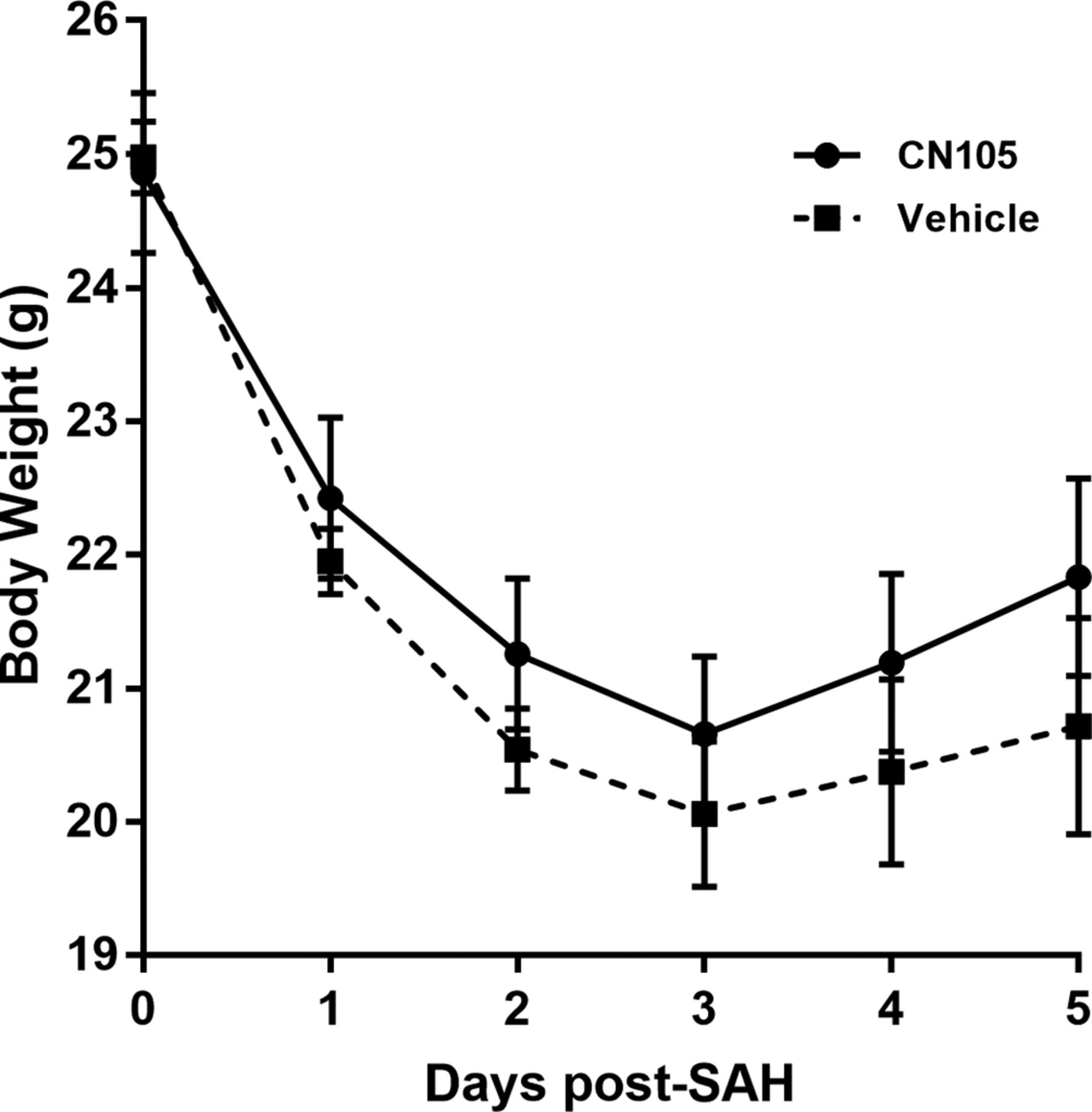
Treatment with CN-105 was associated with a trend towards recovery of body weight following induction of SAH (p=0.052). SAH, subarachnoid haemorrhage.
CN-105 improves functional outcome after SAH
To investigate CN-105 effects on neurological recovery, 0.05 mg/kg CN-105 (n=12) or vehicle (n=14) was administered by tail vein injection every 12 hours for 5 days, with the first dose given at 2 hours following SAH. Daily vestibulomotor function and neurological examination were assessed by RR test and neurological severity scale starting the day prior to SAH and continuing for 5 consecutive days following injury. Treatment with CN-105 significantly improved RR performance as compared with vehicle treatment (figure 3A, p<0.001). Neurological severity scoring mirrored the RR findings. Mice treated with CN-105 attained markedly higher scores over the 5 days of testing compared with vehicle-treated counterparts (figure 3B, p=0.001).

Treatment with CN-105 was associated with durable improvements in vestibulomotor function and significant reduction of neurological deficits compared with vehicle-treated mice over 5 days after SAH, as demonstrated by longer Rotorod latencies (A, p<0.001) and increased Neuroseverity Score (B, p=0.001). SAH, subarachnoid haemorrhage.
CN-105 decreased microgliosis after SAH
To investigate whether administration of CN-105 was associated with a downregulation in glial response to injury, we performed immunohistochemistry with F4/80, a marker of activated microglia, which was assessed by quantitative stereology. At 5 days after SAH, CN-105-treated animals had a significant reduction in F4/80 positive cells (8383±1281) compared with vehicle-treated controls (36 079±10 460; figure 4A–E).

Treatment with CN-105 significantly decreased microgliosis associated with brain injury after SAH. Images of immunostained brain slices with F4/80 for microgliosis, Vehicle (A and C) and CN-105-treated (B and D), mice sacrificed 5 days post-SAH. Quantification by formal stereology for F4/80 positive cells (E) in the whole hippocampus in vehicle or CN-105-treated mice at 5 days after SAH (Vehicle mice 36 079±10 460; CN-105 8383±1281; p=0.036). SAH, subarachnoid haemorrhage.
CN-105 reduced short-term neuronal injury and long-term neuronal survival
To evaluate whether this reduction in microgliosis was associated with neuroprotection, FJB staining was performed to identify injured neurons in areas of the brain that are preferentially susceptible to injury after SAH, including the CA1 and CA2 region of hippocampus. At 5 days after SAH, there was scattered neuronal injury in the cortex and diencephalon, as demonstrated by Fluoro-Jade staining. Treatment with CN-105 was associated with a significant reduction in the number of injured neurons in hippocampal CA1+CA2 regions (figure 5A–E).
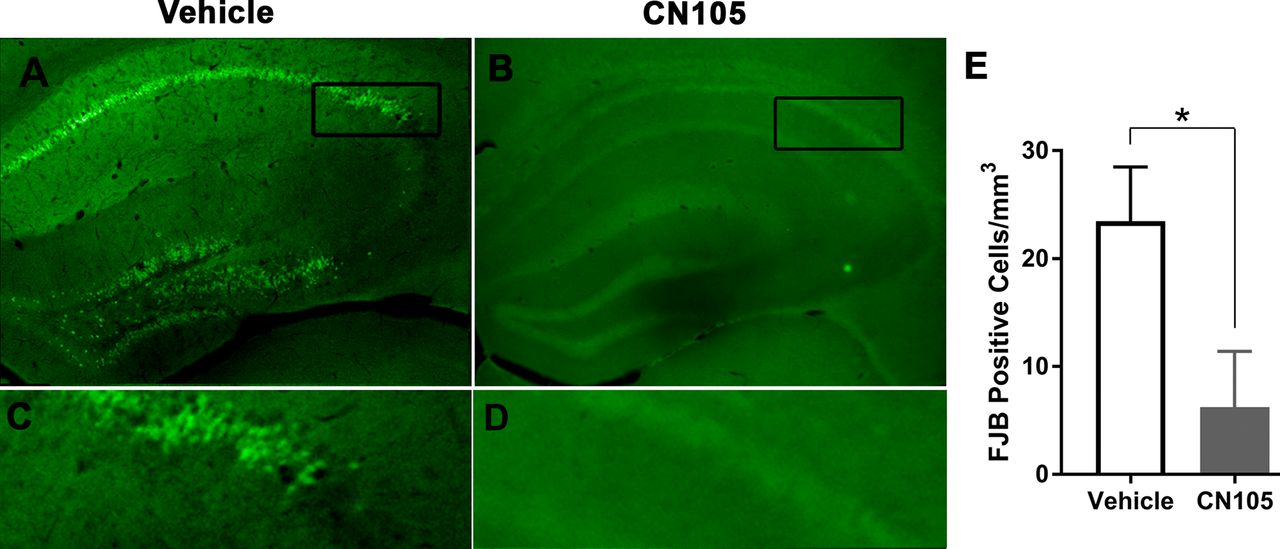
Representative images of FJB positive neurons in the hippocampus. Fluoro-Jade stains degenerating and injured neurons. Prior to injury, there was no evidence of hippocampal Fluoro-Jade staining (data not shown). At 5 days following injury, there were Fluorojade positive hippocampal neurons in mice treated with CN-105 as compared with vehicle (Vehicle 23.47±5.01; CN-105 6.23±5.16; p=0.043). FJB, Fluoro-Jade B.
To assess for longer-term neuronal protection of CN-105, at 35 days after SAH brain slices were stained with NeuN, a neuron-specific marker, and neuronal density was quantified in the CA1 and CA2 regions of the hippocampus. Administration of CN-105 resulted in a robust increase of neuronal density when compared with vehicle-treated animals as assessed by unbiased stereology (figure 6A-C; Vehicle: 132,511±43 955 neurons/mm3; CN-105 273 070±31 527 neurons/mm3, p=0.02).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Representative pictures of NeuN staining performed at 35 days following SAH demonstrate long-term improvements in neuronal viability associated with CN-105 administration. Comparison of NeuN positive cells in the CA1 and CA2 regions of the ipsilateral hippocampus in vehicle-treated animals (A, 132 511±43 955) as compared with mice treated with CN-105 (B, 273 070±31 527) demonstrate a significant improvement in long-term neuronal viability in animals treated with CN-105 (C, p=0.023). SAH, subarachnoid haemorrhage.
Discussion
In the current study, we demonstrate that intravenous administration of CN-105, a five amino acid peptide designed from the aqueous face of the receptor binding region of apoE, was well tolerated in a murine model of SAH and was associated with a reduction in vasospasm as well as improved histological and functional endpoints. Histologically, clinical improvement was associated with a substantial reduction in microgliosis (F4/80 immunopositive cells in ipsilateral hippocampus of CN-105-treated mice at 5 days after SAH). In addition, a reduction in FJB-positive cells and increased density of hippocampal NeuN-positive cells were associated with CN-105-treated mice suggesting that the reduction in microglial activation was also associated with long-term neuroprotective effects. Of note, hippocampal pathology (inflammation and neuronal apoptosis) has been described in a number of preclinical models of SAH33–35 and may serve as the pathological substrate for the cognitive impairment that is commonly observed clinically.36 37 The reduction in microgliosis that we observed suggests that the underlying mechanism of action may be due, in part, to anti-inflammatory effects of CN-105. Although several anti-inflammatory and neuroprotective strategies have been studied to reduce vasospasm and improve functional outcome in this setting, at present no studied pharmacological intervention, with the exception of nimodipine, has ever been demonstrated to improve outcome. The administration of an apoE mimetic therapeutic offers the possibility to address this compelling unmet clinical need.
The model of SAH described in these studies was developed to capture many of the clinical features of aneurysmal SAH associated with acute arterial bleeding into the subarachnoid compartment and is associated with reproducible vasospasm, neuroinflammation and functional deficits.31 In the current study, we demonstrate that the injury was consistent across the different treatment groups. Neuroinflammation has been associated with vasospasm,38 secondary tissue injury and the proliferative arteriopathy associated with vasospasm following clinical SAH, and apoE mimetic compounds have been demonstrated to reduce glial activation and neuroinflammatory responses in vitro and in vivo.18 22 29 39 Our current results are consistent with this proposed mechanism, as treatment with CN-105 was associated with a reduction in luminal narrowing adjacent to the clot and a reduction in microgliosis as assessed by formal unbiased stereology.
The role of endogenous apoE in modulating glial activation and neuroinflammatory responses was first described in 1997,21 40 and data suggest that this effect may be mediated by interaction with the LRP1, a scavenger receptor which is present on microglia and neurons.41–44 This suggested the possibility that the anti-inflammatory effects of endogenous apoE may have therapeutic potential. However, apoE does not readily cross the blood brain barrier and thus has limited therapeutic potential. Creation of smaller peptides derived from the receptor binding region were designed to have increased CNS penetration, and it is believed that binding of synthetic apoE-mimetic peptides to cell surface LRP1 receptors on glial cells45 and neurons initiates signalling cascades that downregulate the inflammatory phenotype, an effect that is not observed in LRP1-deficient microglia.41 46 In prior preclinical models of brain injury, intravenous administration of CN-105 had been associated with a reduction in expressed inflammatory cytokines, microgliosis and cerebral oedema, suggesting that the observed protective effects may be mediated in part by a reduction in inflammation. This is consistent with our current observations demonstrating that CN-105 was associated with a reduction in hippocampal microgliosis.
Although modulation of neuroinflammation may be one mechanism by which CN-105 is associated with durable functional improvement, it is also possible that it exerts direct neuroprotective effects. Neuronal excitotoxicity contributes to tissue injury following SAH, and both apoE and apoE mimetic peptides have been demonstrated to exert direct neuroprotective effects in vitro.47 48 Our current results demonstrating a reduction in subacute hippocampal FluoroJade staining and long-term NeuN neuronal density would suggest that treatment with CN-105 may have direct neuroprotective effects, although indirect effects via modulation of inflammation may play a role as well. Several mechanisms have been proposed to explain the direct neuroprotective effects of apoE and mimetic peptides, including modulation of NMDA receptor trafficking, and indirect modulation of NMDA receptor function via LRP1 interaction with the PSD-95 scaffold protein.48–51
The current results suggest that apoE mimetic peptides may have promise for clinical translation in the setting of SAH. A number of apoE mimetic peptides have demonstrated efficacy in preclinical SAH models.24 25 52 In the current study, we demonstrated the efficacy of CN-105, a smaller and more potent apoE mimetic that has been recently translated into clinical trials.30 CN-105 was developed by ‘linearising’ the polar face of the apoE amphipathic helix involved in receptor interaction. This has the advantage of increased potency and CNS penetration. Our results are consistent with recent reports demonstrating that CN-105 improves functional and histological outcomes in preclinical models representing different mechanisms associated with secondary brain injury, including direct mechanical brain trauma, intraparenchymal haemorrhage and cerebral ischaemia.27–29 Moreover, CN-105 has significant translational potential, given its excellent safety profile and linear pharmacokinetics demonstrated in early phase I clinical trials.30
Although the results of this study are promising, there are several limitations which should be addressed. Although there are several preclinical models of aneurysmal SAH, the translation of neuroprotective agents has been notoriously difficult, and there are no models which have successfully predicted clinical efficacy of a candidate drug. An obvious limitation of all rodent models is the fact that lissencephalic animals may not recapitulate the relevant clinical deficits observed in clinical SAH. Thus, prior to clinical translation, it may be helpful to test efficacy of CN-105 in additional small animal and gyrencephalic models. In addition, it would be helpful to define the effects of gender and comorbidities that would be relevant to early clinical trial design.
In conclusion, we demonstrate that the apoE mimetic peptide CN-105 reduces histological evidence of vasospasm and has a durable effect on improving functional and histological outcomes following SAH. The protective effects of CN-105 are likely due to its effects on dampening maladaptive CNS inflammatory responses and are consistent with its effects in other preclinical paradigms of acute brain injury. Although several anti-inflammatory and neuroprotective strategies have been studied to reduce vasospasm and improve functional outcome in this setting, at present no studied pharmacological intervention, with the exception of nimodipine, has ever been demonstrated to improve outcome. Given its favourable pharmacokinetic profile, CNS penetration and demonstration of safety, CN-105 represents an attractive therapeutic candidate for treatment of brain injury associated with SAH.
References
Footnotes
JL and GZ contributed equally.
Contributors DTL, HW, JL, GZ and BJK designed the study, analysed the data and prepared the main manuscript text and figures. JL, GZ, YT, CF and HW executed the animal modelling, functional tests and histology. All authors reviewed the manuscript.
Funding This work was partially supported by DOD contract CDMRP#W81XWH-16-C-0142.
Competing interests DTL is an officer of Aegis CN, LLC, which supplied study drug. Aegis had no editorial control over study design, execution or the writing of this manuscript. DTL and BJK are coinventors of the Duke-held patent for CN-105.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data sharing will be in compliance with institutional policies.