- 1Department of Neurology, NorthShore University HealthSystem, Chicago, Illinois, USA
- 2Department of Neurology, University of Chicago, Chicago, Illinois, USA
- Correspondence to Dr John H Pula; jpula12009{at}gmail.com
- Received 12 March 2017
- Revised 30 May 2017
- Accepted 2 June 2017
Abstract
A large portion of the central nervous system is dedicated to vision and therefore strokes have a high likelihood of involving vision in some way. Vision loss can be the most disabling residual effect after a cerebral infarction. Transient vision problems can likewise be a harbinger of stroke and prompt evaluation after recognition of visual symptoms can prevent future vascular injury. In this review, we discuss the visual aspects of stroke. First, anatomy and the vascular supply of the visual system are considered. Then, the different stroke syndromes which involve vision are discussed. Finally, topics involving the assessment, prognosis, treatment and therapeutic intervention of vision-specific stroke topics are reviewed.
- Vision
- Stroke
- Amaurosis Fugax
- Homonymous hemianopia
- Diplopia
Anatomy and blood supply
Visual impairment after stroke impacts quality of life and leads to loss of independence and depression.1 2 Vascular occlusion along the afferent or efferent visual pathways can produce myriad effects, including transient monocular vision loss (TMVL), visual field deficits or ocular dysmotility. To understand the variety of stroke syndromes affecting vision, we should first recall the physiological architecture of the visual system in relation to its blood supply.
The prechiasmal visual pathway consists of the axons from the nerve fibre layer of the retina, which forms the optic nerves and then travel medially towards the optic canals. The prechiasmal optic nerves are supplied by ophthalmic artery and internal carotid artery (ICA) pial vessels.1 The central retinal artery, a branch of the ophthalmic artery, provides the blood supply to the retina.3 The optic nerves conjoin to form the optic chiasm, which is supplied by Circle of Willis.1
The retrochiasmal visual pathway encompasses the region from the optic chiasm to the visual cortex.1 Fibres from the optic chiasm and the optic tracts, supplied primarily by the anterior choroidal artery (AChA), travel to the lateral geniculate body (LGB).1 Though the AChA,a branch of the internal carotid artery (ICA)4 and lateral posterior choroidal artery (LPChA),a branch of the posterior cerebral artery (PCA)4 provide dual vascular supply to the LGBs,1 5 6 the terminal anastomosis is vulnerable to ischaemia.7 Optic radiations originate from the lateral geniculate nucleus (LGN) and are divided into superior, inferior, and central nerve fibres. The optic radiations are predominantly supplied by the posterior and middle cerebral arteries1 and the AChA.6 Inferior fibres, known as Meyer’s Loop,6 travel to the temporal lobe, while the superior and central nerve fibre bundles travel to the parietal lobes.1 The termination of optic radiations is located in the visual striate cortex (V1) in the occipital lobe; superior and inferior to the calcarine fissure.1 The occipital cortex is largely supplied by the PCAs, which are the terminal branches off the basilar artery (figure 1).1
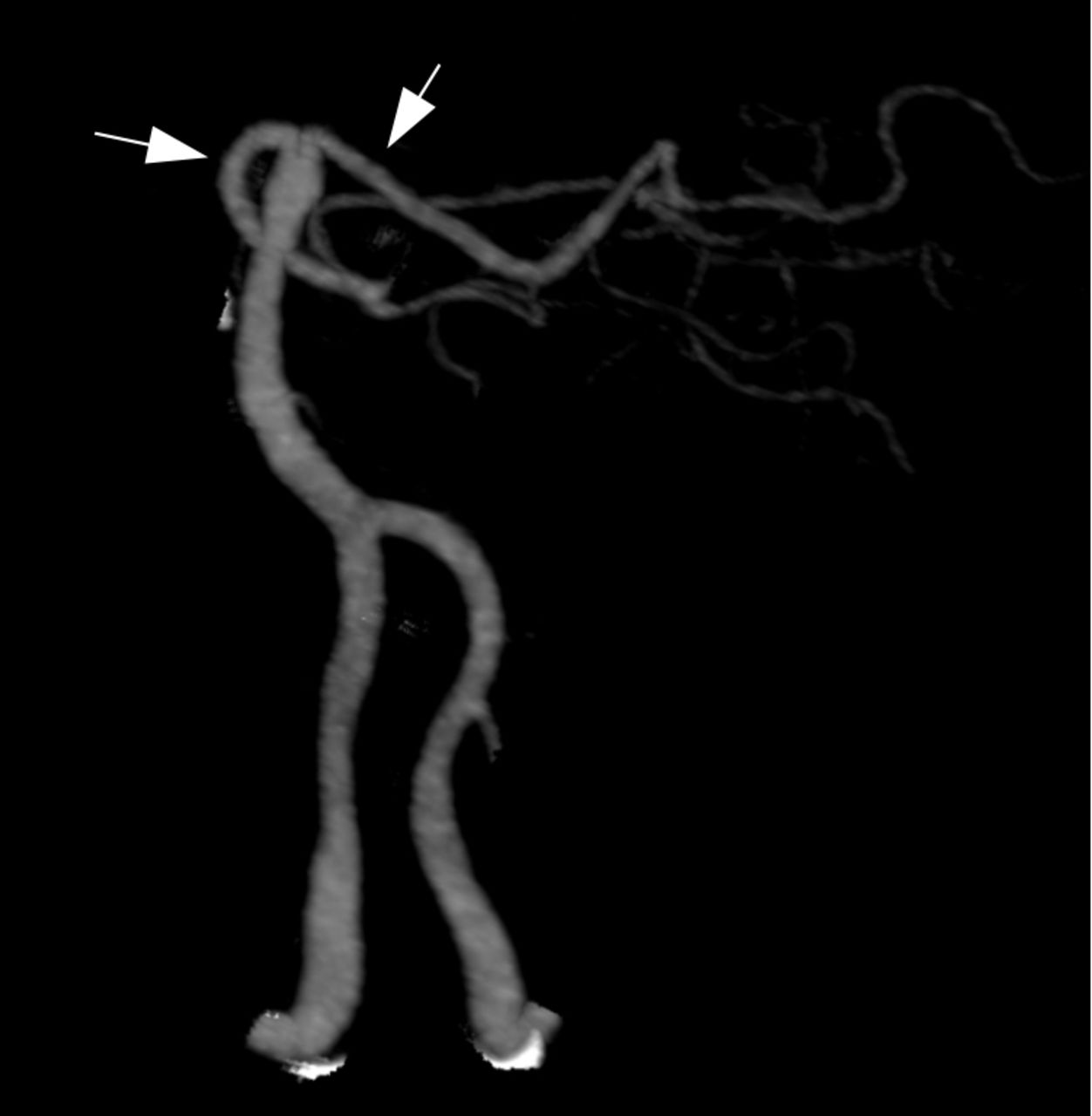
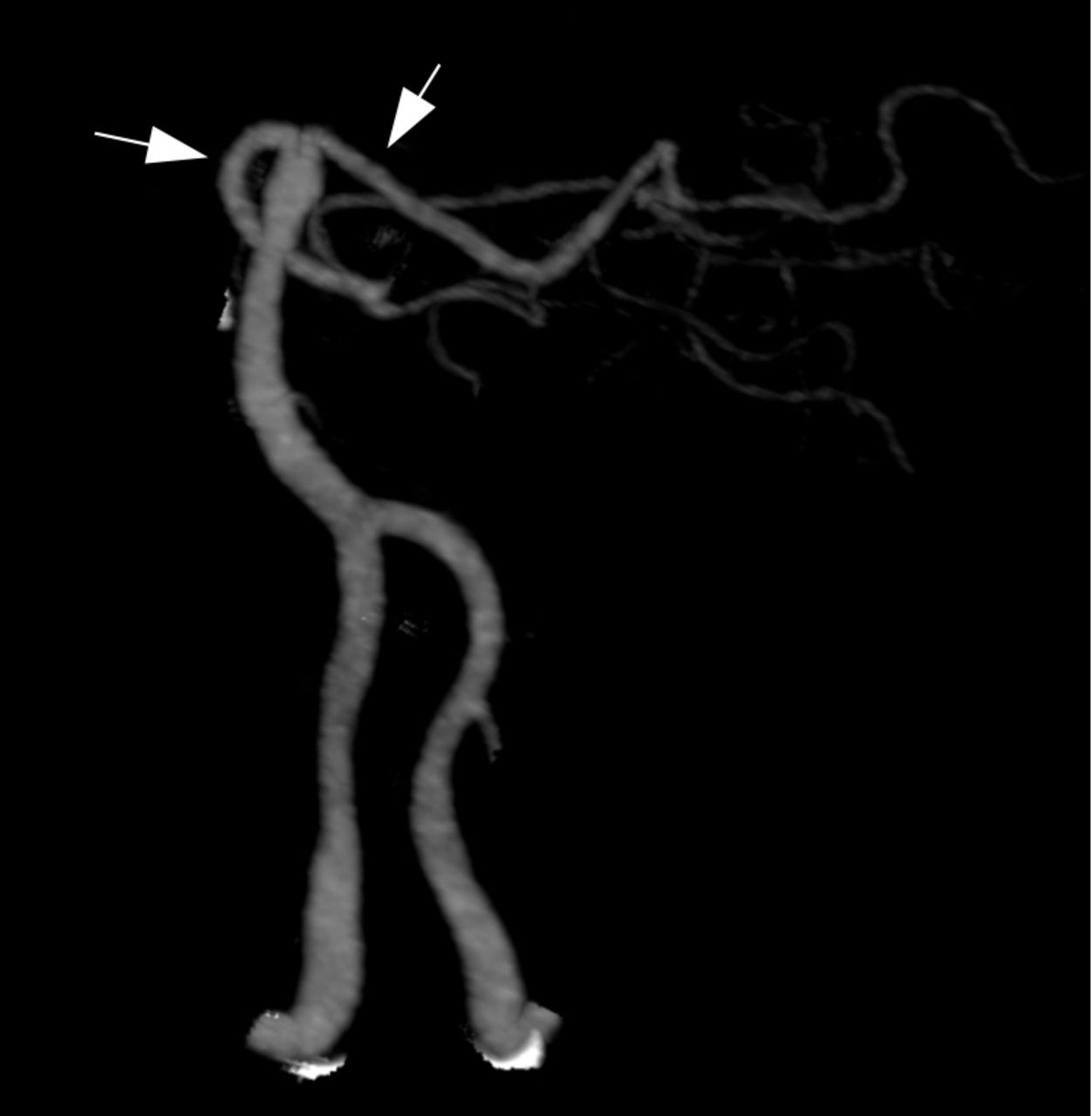
Radiographic angiography of the posterior circulation. Bilateral vertebral arteries fuse to form the basilar artery. The ultimate branches of the basilar artery form the posterior cerebral arteries (PCAs, arrows), which supply the visual cortex. Infarctions in the PCA territories can cause cortical vision loss. Bilateral PCA infarcts as in ‘top of the basilar syndrome’ can result in cortical blindness.
Visual stroke syndromes
The physician can use knowledge of these anatomical landmarks and their corresponding blood supply to clinically localise a stroke involving vision by recognising effects of their dysfunction on the clinical examination.8
Monocular visual loss due to prechiasmal ischaemia
Prechiasmal vision loss can be caused by retinal ischaemia secondary to occlusion within the ophthalmic artery vascular supply.9 Preceding symptoms of non-visual strokes often include TMVL.9 Over 75% of these patients experience associated decreased visual acuity.3 Lavallée et al conducted a prospective cohort study and concluded that ischaemia was the causative factor in 93.3% of transient ischaemic attack (TIA) clinic patients presenting with TMVL.10
Retinal ischaemia can occur either transiently as amaurosis fugax (AF) or permanently due to branch retinal artery occlusion (BRAO), central retinal artery occlusion (CRAO),9 or rarely ophthalmic artery occlusion. Any of these can result in permanent vision loss with decreased visual acuity or visual field deficits.11 CRAO can be recognised by visualisation of the pathognomonic ‘cherry red spot’ in the macula (figure 2).
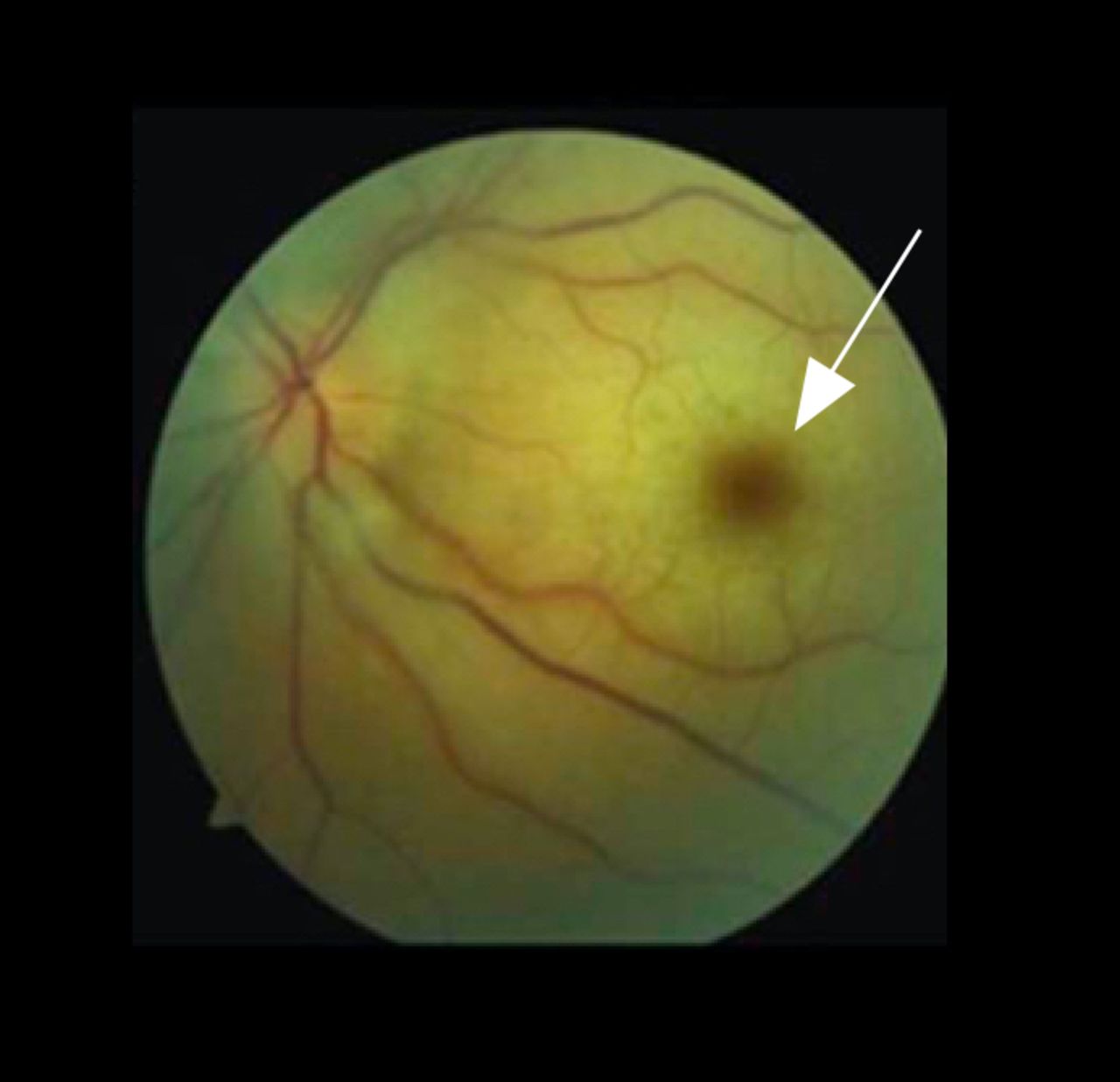
Fundus photo of central retinal artery occlusion (CRAO). CRAO results in generalised retinal oedema, sparing the optic nerve, which is supported by a separate blood supply. Examination shows a ‘cherry red spot’ (arrow). The cherry red spot is an optical illusion. The macula is not more red; the oedema of the nerve fibres surrounding it colours it pallid, causing the macula, which has relatively less nerve fibres and therefore less tissue to swell, to appear relatively red.
Retinal ischaemia is included in the definition of TIA, according to The American Heart Association/American Stroke Association guidelines.12 13 The 5-year incidence of retinal emboli is 0.9% and the prevalence is 1.3%, according to the Beaver Dam Eye Study (BDES), a large population cohort study of 4926 patients.14 The BDES found that asymptomatic patients with retinal emboli experienced an increased risk of stroke-related death (Hazard Ratio (HR)=2.61, 95% Confidence Interval (CI) 1.12 to 6.08) compared with those without retinal emboli.14 Lauda et al performed a retrospective study and concluded that acute brain infarction occurred in 23% of 213 patients with CRAO or BRAO, a significant finding given that vision loss was the only presenting symptom in 90% of these patients.9
Transient binocular visual loss (TBVL) is not due to a prechiasmal occlusive process, but rather vertebra-basilar ischaemia.11 TBVL produces homonymous visual field defects and is as much a warning sign of stroke as is atrial fibrillatin (AF).11
Bitemporal hemianopia due to chiasmal ischaemia
Chiasmal strokes are rare, owing to the rich supply of collateral circulation provided by the Circle of Willis to the optic chiasm.15 16 When chiasmal strokes do occur, patients experience acute onset bitemporal hemianopia (figure 3). Atypical presentations can also occur, such as Shelton et al reporting a case of right temporal visual loss with complete contralateral monocular vision loss, the so-called junctional scotoma.15 Subsequent imaging revealed an anterior chiasmal infarction.15 Fabian et al cite the case of a patient who presented with a junctional scotoma resulting from a right chiasmal and optic nerve infarction following aortic valve replacement.16

Humphrey visual field showing bitemporal hemianopia. Visual fields are represented with the left eye on the left and right eye on the right, which is opposite to how CT or MRI is viewed. Therefore, the temporal fields are laterally represented. Bitemporal hemianopia will generally respect the vertical meridian and imply the chiasm as the location of the insult.
Homonymous hemianopia due to postchiasmal ischaemia
Postchiasmal strokes occur secondary to ischaemia in the LGB, optic radiations, or occipital lobe and can manifest as sectoranopias, quadrantanopias, or hemianopias, either congruous or incongruous.
Incongruous visual field loss due to optic tract and lateral geniculate body infarction
Incongruous visual field defects can arise from optic tract and LGB ischaemia.4 6 Incomplete injury to the optic tract and the spatial spread of visual fibres in the optic tract can lead to incongruous contralateral homonymous hemianopia (HH).6 A more distinctive pattern of sectoral optic atrophy with resulting ipsilateral temporal disc pallor and contralateral wedge-shaped pallor can also be observed.6 17
The LGN comprised six layers.4 Ipsilateral retinal information is processed in layers 2, 3 and 5.4 Contralateral retinal information is processed in layers 1, 4 and 6.4 The parvocellular layers (3, 4, 5 and 6) mediate colour perception.4 6 The lateral geniculate body’s location in a watershed area increases ischaemic vulnerability during periods of hypoperfusion.18 Extensive LGN injury manifests as a complete HH.6 Medial LGB infarction manifests as a wedge-shaped HH.5 A hemi-hourglass shape in the horizontal midline of the visual field is a consequence of horizontal wedge-shaped homonymous sectoranopia.6 Tsuda et al reported a case of an 81-year-old with diabetes presenting with left incongruous wedge-shaped HH secondary to right LGB infarction.5 de Vries et al report the case of an 89-year-old woman presenting with right visual field symptoms of golden coloured balls with green borders, with subsequent development of a grid-like pattern consisting of black dots arranged symmetrically, both horizontally and vertically.4 Subsequent MRI demonstrated a subacute LGN and ventrolateral thalamic lacunar infarct.4
Visual disturbance induced by bilateral LGB infarction is a rare occurrence.10 Silva et al cite a case of a 10-year-old girl presenting with field loss due to severe diarrhoeal induced hypoperfusion.18 MRI demonstrated bilateral LBG haemorrhagic lesions.18 Lefèbvre et al also describe a case of bilateral LGB infarction secondary to hypoperfusion in a 31-year-old woman who experienced amoxicillin-induced anaphylactic shock.7 She presented with incongruous bitemporal and binasal visual field deficits with acute bilateral vision loss and dyschromatopsia.7
Incongruous HH or quadrantanopia due to optic radiation infarction
Anterior optic radiation lesions can cause incongruous contralateral homonymous hemianopic or quadrantanopic visual field deficits with sloping borders and associated hemiparesis.6 Complete HH is produced with occlusion of both LPChA and AChA.4
Quadrantanopia is visual loss occurring in the identical quadrant of the visual field binocularly. Combined superior or inferior quadrantanopia, with horizontal meridian sparing, results from proximal AChA occlusions.4 Exclusive superior or inferior quadrantanopia results from AChA branch occlusions.4 Contralateral homonymous superior quadrantanopia, so-called ‘pie-in-the-sky’ defects,6 occurs with temporal lobe infarction involving Meyer’s Loop19 with possible associated symptoms of seizure, memory deficits and receptive aphasia.6 Contralateral homonymous inferior quadrantanopia (pie-on-the-floor) occurs with parietal lobe infarction, involving the parietal optic radiations19 with associated hemineglect.6
Congruous homonymous hemianopia due to occipital lobe infarction
Up to 8%–25% of patients who had a stroke can develop visual field loss.1 Stroke is the most common causative factor for HH19 and correspondingly, HH is the most common form of visual field loss following stroke.20 Rowe et al performed a prospective multicentre cohort study consisting of 915 patients who had a stroke and concluded that complete HH (54%) represented the most frequent type of visual field loss, followed by partial HH (19.5%), superior or inferior quadrantanopia (15.2%), visual field constriction (9.2%), scotomas (5.1%) and bilateral hemianopia/cortical blindness (1.7%).1 Another large retrospective review of cases had similar findings with HH representing largest percentage of cases (69.7%).19 A third retrospective study assessed 220 patients with either traumatic brain injury (TBI) or cerebral vascular accident and also concluded that HH (47.5%) had the greatest frequency of occurrence in visual field defects, with scattered and non-homonymous defects each representing 20% of visual field defects.21
Congruous HH is predominantly caused by occipital lobe lesions (54%), followed by the optic radiations (33%), optic tract (6%), LGN (1%).1 One large retrospective review correspondingly found that occipital lobes and optic radiations also defined the most commonly affected regions at 45.0% and 32.2%, respectively.19
PCA ischaemia can result in congruous occipital lobe lesions (figure 4).19 Increased congruity occurs with lesions occurring more posterior in the visual pathway due to convergence in the occipital lobe.6 Occipital lobe lesions can cause field defects which have macular sparing, where the centre of the visual field remains unaffected. The dual blood supply from the PCA and the superior temporo-occipital sylvian artery is one reason for the macular sparing.19

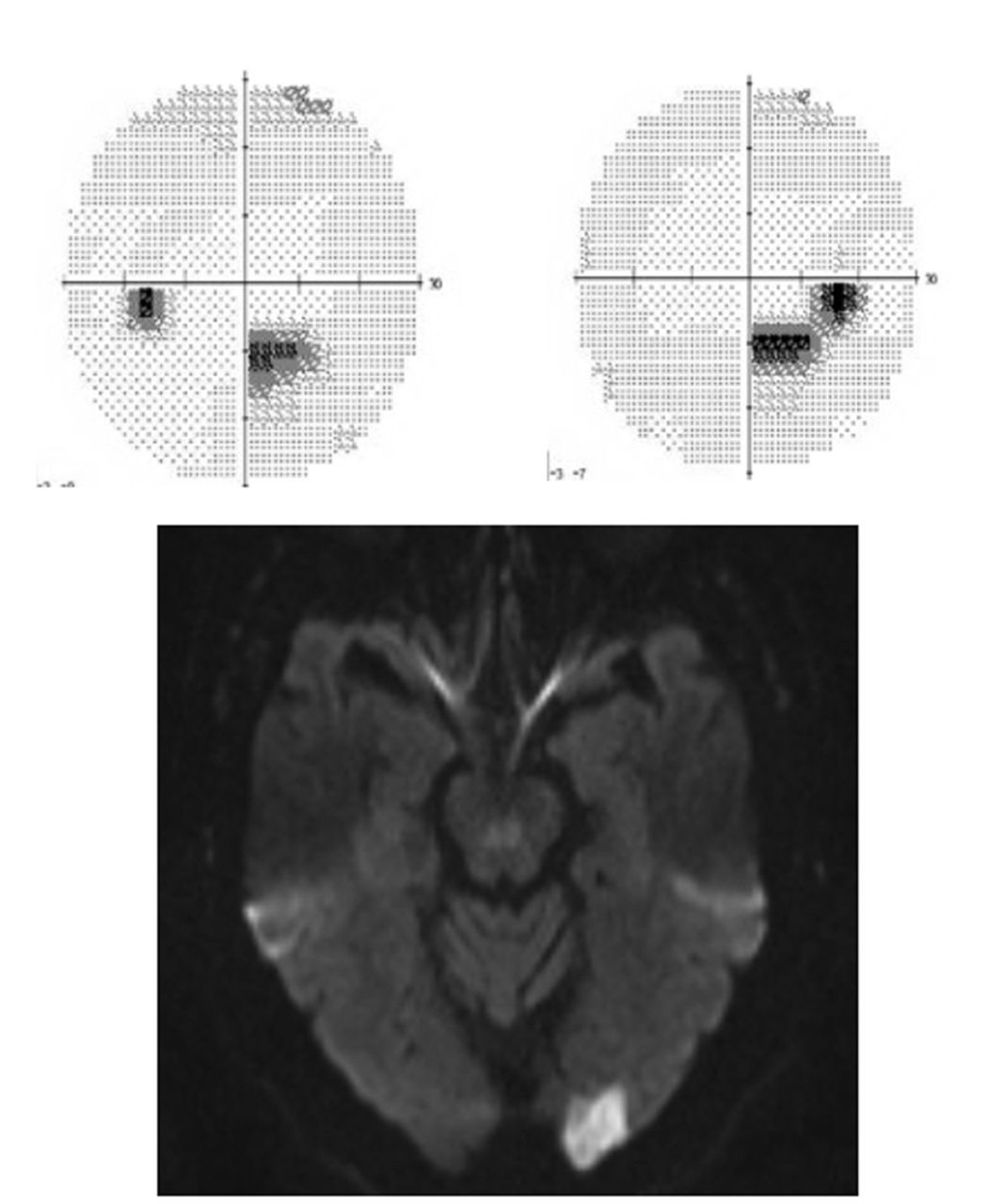
Visual fields and diffusion-weighted imaging MRI showing occipital infarction. Humphrey visual fields (above) show a partial congruous right inferior field defect. Diffusion-weighted MRI (below) shows an acute infarction of the left occipital pole. The visual fields correlate in terms of localisation to the radiological defect.
Cortical blindness due to bilateral occipital lobe infarction
Cortical blindness is indicative of bilateral occipital lobe ischaemia.19 Associated agnosia of the cortical blindness is known as Anton syndrome.19 Intact pupillary light reflex and normal appearing fundi are key diagnostic elements of cortical blindness which localise the lesion to the posterior visual pathway.19
Efferent visual dysfunction due to lesions outside the afferent visual pathways
Other forms of visual impairment due to lesions external to the visual pathway include ptosis, diplopia, internuclear ophthalmoplegia (INO), one-and-a-half syndrome, gaze palsies, saccadic intrusions, impaired smooth pursuits, and nystagmus.
Ptosis
Pure midbrain infarctions represent a small number of strokes (0.6%–2.3%).22 Paramedian midbrain infarctions can manifest as ipsilateral ptosis.22 Oculomotor fascicular infarction can also induce isolated unilateral ptosis without ophthalmoplegia or pupillary constriction restriction.22 Sugawara et al report a case of isolated left-sided ptosis without ophthalmoplegia and vertigo in a 59-year-old man with hypertension, diabetes mellitus, dyslipidaemia,and glaucoma.22 MRI revealed an acute left paramedian midbrain infarction.22
Acute onset bilateral ptosis involves both levator palpebrae superioris implying injury to the unpaired midline central caudal nucleus.8 22 The central caudal nucleus, a subnucleus of the oculomotor nucleus in the midbrain, mediates the bilateral levator palpebrae superioris muscles.22 Khandker et al describe a case of acute onset horizontal diplopia and bilateral ptosis in a 69-year-old man with hypertension, uncontrolled diabetes mellitus, dyslipidaemia, and macular degeneration.8 Examination revealed bilateral ptosis and a left INO.8 MRI revealed findings consistent with subacute ischaemic stroke involving the oculomotor nerve nucleus and the medial longitudinal fasciculus (MLF).8
Ocular dysmotility
A high number of patients who had a stroke develop ocular dysmotility or alignment impairment,23 presenting as diplopia, INO, one-and-a-half syndrome, as palsies/paresis including gaze, saccadic or smooth pursuit.24–26 One retrospective study reviewed 220 records of patients with stroke or traumatic brain injury (TBI) to determine the oculomotor impairment rate and concluded strabismus and cranial nerve III palsy had the highest rate of occurrence.27
Oculomotor nerve injury is characterised by ptosis, gaze palsy, and mydriasis due to denervation to the levator palpebrae superioris, ciliary body, pupillary constrictors, superior rectus, inferior rectus, medial rectus, and inferior oblique extraocular muscles.8 Patients with stroke who have a oculomotor palsy often have other neurological findings such as hemiparesis or cerebellar dysfunction. An isolated oculomotor nerve palsy often implies a different mechanism, such as compression or diabetic microvascular injury. When an oculomotor nerve palsy is caused by damage involving the corticospinal tract prior to decussation of fibres in the medulla oblongata, the resultant combination of ipsilateral third nerve palsy and contralateral hemiparesis is known as Weber’s Syndrome, which localises to infarction in the midbrain. A different mechanism of oculomotor palsy, which is vascular but compressive, is aneurysm of the posterior communicating (PComm) artery. An expanding aneurysm in the PComm produces compression to the oculomotor nerve. Because the pupil fibres are external to the ocular motor fibres, compression results in anisocoria with a mydriatic (enlarged) pupil either prior to or concomitant to diplopia and ophthalmoparesis. This is the so-called ‘rule of the pupil’. In contrast, microvascular third nerve palsy, which affects the innermost motor fibres preferentially prior to the pupil fibres, should not cause a ‘blown pupil’ or result in significant anisocoria out of proportion to the motor deficits.
Diplopia equally occurs in right versus left-sided strokes.23 Diplopia can be a consequence of horizontal or vertical ocular misalignment,26 either from third, fourth, or sixth cranial nerve palsy (figure 5) or from skew deviation.28 A prospective multicentre observational case cohort study by conducted by Rowe et al found that 16.5% of patients with poststroke had ocular misalignment with diplopia.23

Sixth nerve palsy. In this image, the patient is asked to look right. His right eye does not abduct, while his left eye adducts normally. This is typical of an abducens (sixth nerve) palsy.
Impaired horizontal conjugate gaze
Horizontal gaze paresis is a finding indicative of pontine sixth nerve nucleus injury involving the horizontal gaze centre, the parapontine reticular formation (PPRF).26 Bilateral horizontal gaze palsy is rare in occurrence and characteristic of tegmental pontine infarctions.29
INO is predominantly unilateral and characterised by impaired horizontal conjugate gaze with ipsilateral adduction impairment and concomitant contralateral horizontal gaze-evoked abducting nystagmus. Convergence is spared in INO and is a key diagnostic element in discerning INO from a third nerve palsy.8 In patients in their sixth decade and greater, a stroke to the MLF is the most common cause of INO.8
One-and-a-half syndrome, first described by Fisher in 1967,29 is characterised by an ipsilateral horizontal gaze palsy and contralateral adduction restriction.29–31 Eight-and-a-half syndrome, first described in 1998 by Eggenberger, is characterised by one-and-a-half syndrome and ipsilateral facial palsy.29 Eight-and-a-half syndrome is caused by lesions in the dorsal tegmentum in the caudal pons involving PPRF, MLF (one-and-a-half syndrome) and the facial nerve nucleus and fasciculus (ipsilateral facial palsy).32 Bocos-Portillo et al describe a case of an 81-year-old man with hypertension, diabetes mellitus and dyslipidaemia who presented with left INO, horizontal left gaze palsy and left facial palsy.32 Diffusion-weighted imaging (DWI) was consistent with pontine dorsal tegmentum ischaemic stroke.32 Felicio et al report a case of a 73-year-old man with diabetes who was found to have bilateral horizontal gaze palsy and associated right peripheral facial hemiplegia.29 MRI revealed diffusion restriction in the midline pontine tegmentum.29 Utku et al describe a case of a 43-year-old woman with bilateral horizontal gaze palsy and bilateral facial hemiplegia.33 Subsequent MRI demonstrated a mid-pontine paramedian tegmental lesion.33
Gaze defects
Impaired gaze holding can manifest as a result of injury to the interstitial nucleus of Cajal, nucleus prepositus hypoglossi or the medial vestibular nuclei.26 Complete gaze palsy can result from dorsal caudal pontine or bilateral paramedian midbrain–thalamic infarcts.26 Complete vertical, upgaze and downgaze palsies are findings indicative of bilateral rostral interstitial MLF (riMLF) lesions.31 Isolated downgaze palsy can occur from bilateral mediocaudal riMLF lesions, whereas isolated upgaze palsy can occur from unilateral mesenphalic reticular formation lesions.31
Vertical gaze is facilitated by the third and fourth cranial nerves and is impaired in midbrain and thalamic infarcts.26 Parinaud’s syndrome can arise from dorsal midbrain strokes and is characterised by impairment of upward saccades and pursuit, light-near dissociation and convergence-retraction nystagmus.26 30 Lid retraction, known here as Collier’s sign, is also associated with Parinaud’s syndrome.30
Saccades
Voluntary horizontal saccades are volitional rapid eye movements usually mediated by the frontal lobes. Both cortical and brainstem infarcts can lead to saccadic defects.26 The left frontal lobe facilitates a horizontal saccade to the right with right lateral rectus and left medial rectus activation.30 A lesion to the left frontal lobe results in transient right gaze palsy and left gaze preference, which resolves when the PPRF assumes control for saccades.30 Hypermetric saccades towards the affected side and concomitant hypometric saccades towards the contralateral side can occur in infarction in the brainstem and cerebellum.31
Impaired smooth pursuits
Pursuits are visual tracking of slower moving targets.30 Impaired smooth pursuit movements can be caused by ipsilateral occipitoparietal lesions.30 The right occipitoparietal region mediates a right horizontal pursuit movement with right lateral rectus and left medial rectus activation.30 Simultaneous saccadic palsy and impaired smooth pursuit can be caused by ipsilateral PPRF lesion.30 Cerebellar infarctions involving the vermis, specifically in the uvula (lobule IX) and the pyramid of the vermis (lobule VIII), can also impair smooth pursuits.34 35 Pierrot-Deseilligny et al report a case of an 80-year-old woman who presented with impaired smooth pursuits.36 MRI revealed infarction in the cerebellar vermis, lobules VI to X).36
Nystagmus
Bidirectional horizontal nystagmus can occur in posteroinferior cerebellar strokes with greater amplitude toward the ipsilateral side of the lesion.31 Upbeating nystagmus can occur in cerebellar strokes involving the superior vermis.31
Assessment of visual defects in stroke
Vision problems are often under-reported in patients who had a stroke. When they are recognised, prognostic outcome is dependent on the self-reported symptoms as well as ancillary test performance.24 It is imperative to investigate self-reported symptoms because stroke can cause visual symptoms without objective quantifiable visual impairment signs.24 Correspondingly, patients who had a stroke without visual symptoms were at times found to have objective visual impairment.24 Rowe et al performed a multicentre prospective study of 799 visual assessment referrals from multidisciplinary stroke teams and concluded that 92% of patients who had a stroke had visual impairment despite the fact that only 42% of multidisciplinary stroke teams reported objective findings of ocular impairment.37 In cases of absent ocular impairment, referrals were based on the suspicion of visual difficulty, such as neglect, closing one eye and head compensation.37
Screening for visual problems in stroke
Screening includes standardised visual and eye motility assessment. Existing screening methods for poststroke visual impairment do not provide comprehensive evaluation of visual impairment and furthermore, lack sensitivity due to reliance on self-reported symptoms.38 The National Institutes of Health Stroke Scale (NIHSS), Rapid Arterial oCclusion Evaluation Scale and Texas Stroke Intervention Pre-hospital Stroke Severity Scale screening tools include visual field loss as a component.39 However, the Los Angeles Motor Scale, Hemiparesis, 3-Item Stroke Scale and Cincinnati Pre-hospital Stroke Severity Scale do not consider visual field loss in their assessment.39 Teleb et al developed a novel screening tool for emergent large vessel occlusion, termed Vision, Aphasia and Neglect (VAN) and includes visual field testing as a factor in the screening process.39 The authors concluded VAN was 100% sensitive and 90% specific in their single centre study consisting of 62 stroke code activations.39
Hanna et al performed a systematic review to determine the efficacy of screening tools for visual impairment poststroke, including NIHSS, Functional Impairment Battery, Occupational Therapy Perceptual Screening Test, The Sunnybrook Neglect Assessment, Line Bisection and Text Reading.38 The authors concluded that the existing tools provided incomplete evaluation of poststroke visual impairment and further recommended research and development of a single standardised comprehensive tool for poststroke visual impairment.38
Retinal ischaemia as a screening indication for neuroimaging for stroke
The American Heart Association/American Stroke Association (AHA/ASA) guidelines recommend immediate neuroimaging for patients with retinal ischaemia,40 given that it may be one of the predictors of stroke.12 Neuroimaging is performed to screen for concomitant cerebral ischaemia. Underlying stroke risk factors should be investigated, including diabetes mellitus, hypertension, hyperlipidaemia, coronary artery disease and tobacco use.28 DWI is recommended by Lee et al for patients with retinal ischaemia.41 Lavallée et al concluded that 20% of their patients with transient ischaemic visual symptoms had underlying risk factors for stroke recurrence.10 The authors also found that approximately one-third of their patients with possible or definitive TIA or stroke and concomitant lateral HH also had risk factors for an embolic source.10 Twenty-three per cent of patients with lateral HH were found to have atrial fibrillation, 2.5 times greater than that in patients without transient visual symptoms and more than six times greater than patients presenting with other transient visual symptoms.10 Lauda et al found that 18.2% of retinal ischaemia patients experienced recurrent vascular ischaemic at 1-month follow-up.9 Helenius et al found that 71% of patients with TMVL had simultaneous silent brain infarction.40 The BDES advocate further workup for hypertensive control and possible prevention of stroke-related death asymptomatic hypertensive patients with retinal emboli.14
Carotid artery stenosis is an underlying risk factor for stroke and occurs 4.1% in the general population.42 The European Assessment Group for Lysis in the Eye (EAGLE) Study, a randomised, controlled, prospective multicentre study consisting of 84 patients, found that 40% of their patients with CRAO had ipsilateral carotid artery stenosis of 70% or greater.42 The authors thereby recommend carotid imaging for patients with CRAO.42
Evaluation of acute isolated ocular motor palsies diagnosis
No definitive recommendations exist for diagnostic evaluation of acute isolated ocular motor palsies. While acute isolated ocular motor palsies can be seen following a stroke, other aetiologies such as giant cell arteritis, neoplasms, inflammation and pituitary apoplexy can also produce the same symptomatology.28 Some prospective studies advocate early MRI in older patients with acute isolated ocular mononeuropathies, while other studies support a 3–6-month period of observation alone due to low yield, cost and indeterminate adverse outcome.28 In the absence of spontaneous resolution following the observation period, MRI is more definitively warranted.28
Treatment
Visual field deficits are associated with an increased risk of falls and institutionalisation, impaired reading ability1 and rehabilitation outcome.2 20 Visual field defects are also associated with poor clinical outcome43 and mortality.2 Kim et al found that diplopia (OR=0.26, p=0.023) and visual field defects (OR=4.87, p=0.014) were independent predictors of worse clinical outcomes in patients with moderate-to-severe posterior circulation stroke, as measured with 3-month modified Rankin Scale of 3–6.43
Visual field loss interventions
Poststroke visual impairment interventions can be categorised into deficits involving ocular dysmotility, visuospatial deficits, perceptual deficits44 and the most common, field loss.45 No definitive standard treatment options exist due to insufficient high-quality evidence-based research in this area.44 45 Spontaneous recovery can occur with varying degrees of visual field enlargement1 and it predominantly occurs within the first 3 months following stroke.44 Millington et al concluded that HH occurs in 8% of patients who had a stroke with spontaneous recovery in 50% of patients who had a stroke.46 Zhang et al reported spontaneous improvement of visual field impairment in 38.4% of their patients with HH.47 For patients who do not achieve spontaneous resolution, HH treatment modalities include eye movement training, optical and restorative therapies.46 Visual prognosis using Vision Restoration Therapy and Neuro-Eye Therapy is dependent on retrograde degeneration.46 Occipital lobe damage can lead to optic tract degeneration.46 Optic tract axonal injury leads to retrograde degeneration of retinal ganglion cells.6 11 Millington et al concluded that optic tract degeneration was proportional and significantly correlated to acquired occipital lobe damage (r=0.95; p<0.005; one-tailed Pearson correlation).46
Ocular dysmotility interventions
There are multiple interventions for the management of ocular dysmotility poststroke, including restorative, compensatory and substitutive measures.25
Restitution aims to restore visual function by repetitive convergence, saccadic and pursuit stimulation training.25 The AHA/ASA evaluated eight studies that assessed ocular dysmotility, visual field deficits and perceptual deficits and found that evidence for restitutive therapy efficacy was largely inconclusive.44 However, there is evidence to support the use of eye training for convergence insufficiency.44
Compensatory measures target the disparity between the patients’ deficits and the abilities required to perform the daily tasks by eye movement training and compensatory head posture and movement.25 Though compensatory scanning measures may improve visual field, scanning and reading outcomes, evidence is lacking in improvement of activities of daily living.48 However, there is evidence to support the use of visual rehabilitation and computer-based treatment for visual field defects.44 Mödden et al performed a randomised study of 45 inpatient rehabilitation patients with visual field deficits and determined that concluded that computer-based compensatory therapy improved functional deficits over standard occupational therapy.49
Substitutive measures employ devices that alter visual images through utilisation of magnifiers, prisms and patches.25 Pollock et al performed a Cochrane systematic review to determine the efficacy of ocular dysmotility intervention poststroke and report that the results were inconclusive and recommend future randomised controlled trials.25 Barrett conducted literature review of behavioural optometry and found a lack of evidence-based controlled trials.50 Fresnel prism segments (Peli lenses) have been purported to improve visual fields in patients with HH by increasing perceived direction.
Some physicians support the use of yoked prisms for visual field deficits.44 Prisms, complete occlusion, sectoral occlusion,23 26 orthoptic exercises have all been used as treatment for diplopia.23 Rowe et al recommend orthoptist referral during acute stroke and neurorehabilitation to evaluate and manage strabismus and diplopia, given that approximately 50% of patients who had a stroke have no complaints of diplopia and may be missed.23 Ocular motility surgery or botulinum toxin can be considered as well for diplopia.26
Perceptual deficits interventions
Visual organisation skills systematic training may be considered for perceptual deficits in the absence of visual neglect following right hemispheric stroke.44
CRAO interventions
Treatment for acute CRAO caused by atherosclerotic disease include both non-invasive management and invasive procedures.3 Non-invasive treatment includes for oxygen restoration via retinal arterial vasodilation, embolic dislodgement with ocular massage and improvement in retinal perfusion pressure via reduction of intraocular pressure with acetazolamide or mannitol.3 Invasive treatment with thrombolysis has to be delivered intravenously or intra-arterially.3 Johns Hopkins Hospital conducted a single-centre, non-randomised interventional study and concluded that thromboembolic CRAO treated with local intra-arterial fibrinolytic therapy leads to improved visual prognosis with few complications in patients compared with standard therapy alone;51 however, thrombolysis did not conclusively benefit patients with CRAO, based on the large EAGLE trial.42
Pathognomonic ocular examination findings
Some aspects of the physical examination of the orbit or fundus aid in the diagnosis neurovascular disease. For example, a Hollenhorst plaque in the retina denotes a thromboembolic predisposition. It is a cholesterol plaque appearing yellow or white in the lumen of a retinal artery (figure 6).52 The affected patient may note a visual field defect, or it may be asymptomatic. A workup for vascular disease due to increased future stroke risk is usually obtained.

Hollenhorst plaque. Fundus photo showing a chalky-coloured cholesterol plaque in the superior retinal vascular arcade.
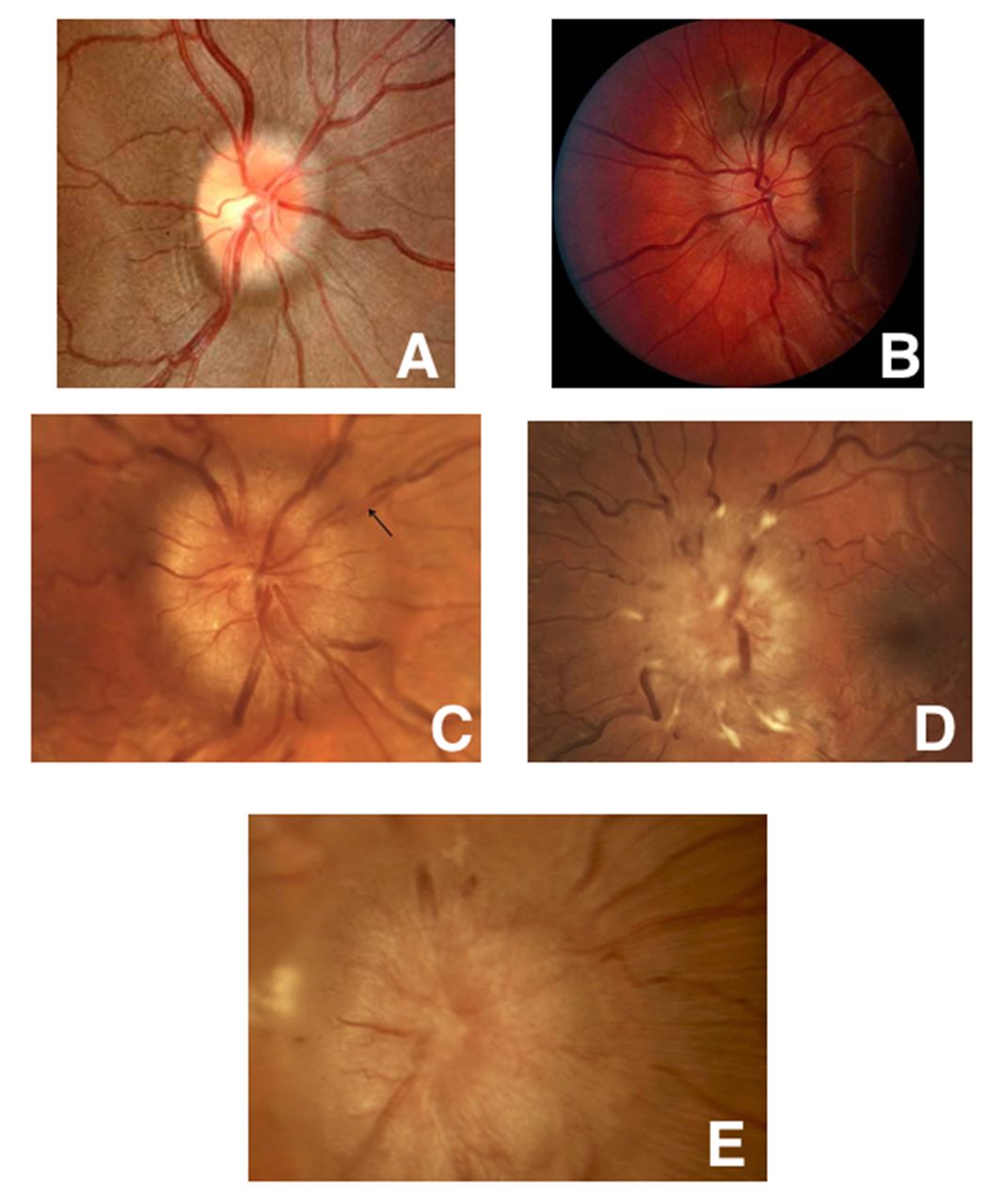
Papilloedema is disc oedema caused by increased intracranial pressure. Although it is not usually vascular in origin, papilloedema can result from intracranial haemorrhage or sequela of the brain oedema secondary to stroke. Papilloedema can be graded on a scale of 1–5 based on certain funduscopic characteristics (figure 7).53
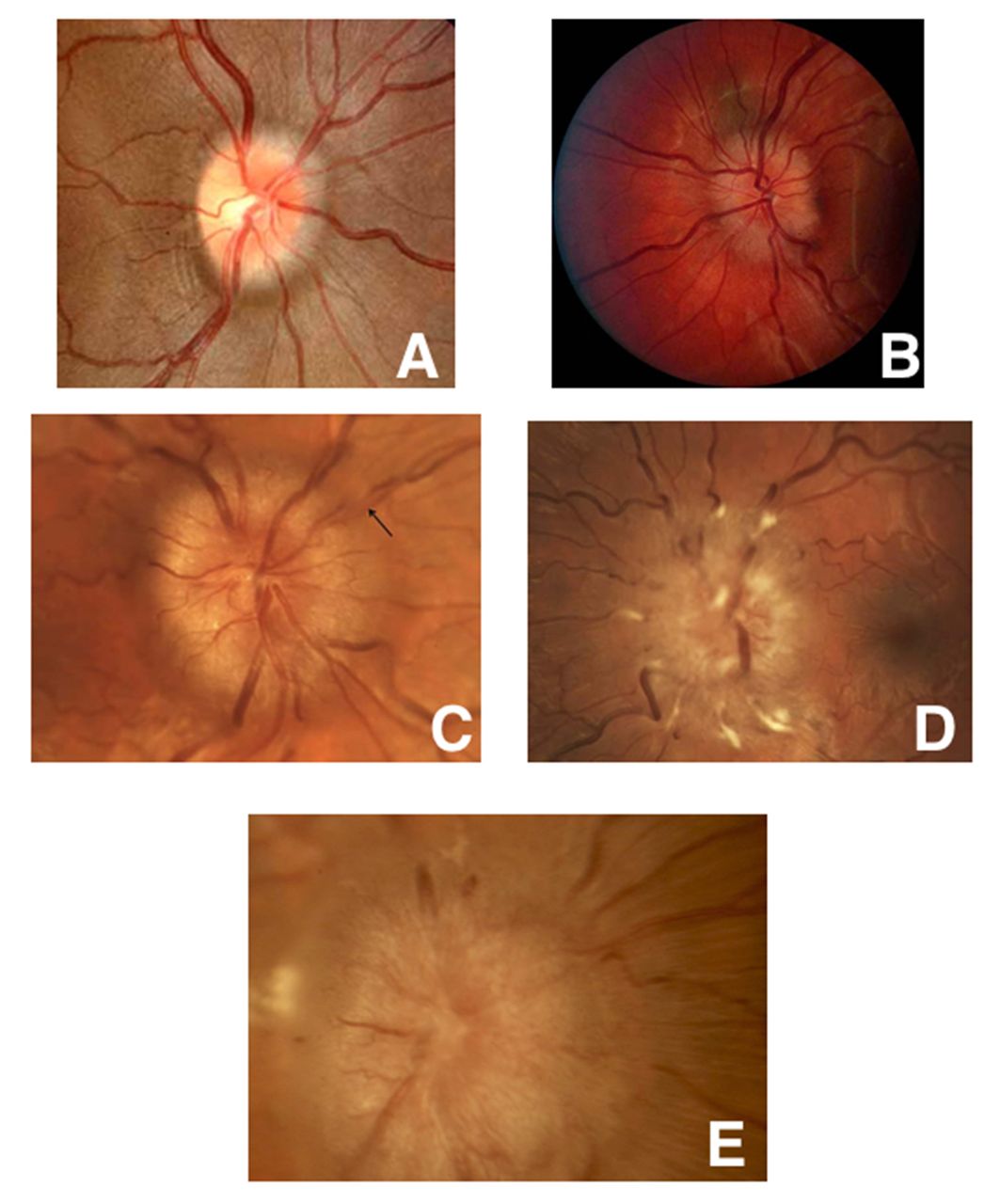
Frisén stages of papilloedema. Five fundus photos showing the various stages of papilloedema. (A) Grade 1: Obscured nasal disc border. (B) Grade 2: Obscuration of all borders with halo. (C) Grade 3: Obscuration of a segment of blood vessel. (D) Grade 4: Total obscuration of one of the blood vessels on the disc head. (E) Grade 5: Dome-shaped protrusion with obscuration of vessels of the entire optic disc.
Posterior pole haemorrhages occurring as a consequence of subarachnoid or intracranial haemorrhage is known as Terson’s syndrome. The pathophysiological mechanism is the rise venous pressure due to stasis from increased intracranial pressure.54 Terson’s syndrome can manifest as a vitreous or preretinal haemorrhage (subhyaloid haemorrhage). The latter type of haemorrhage obscures the underlying retinal vessels and produces a sharply demarcated, boat-shaped appearance due to settling of the blood (figure 8).55

Terson’s syndrome. Terson’s syndrome showing a subretinal haemorrhage after an acute intracranial haemorrhage.
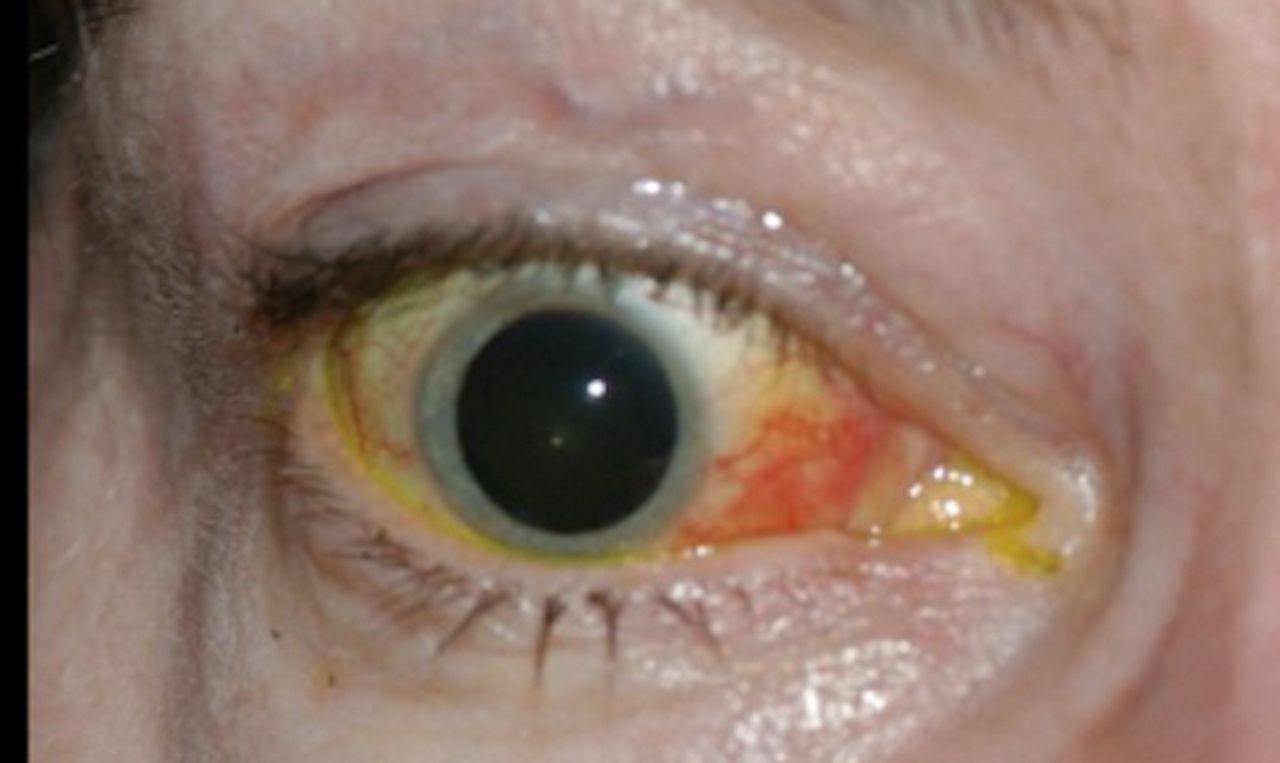
Cavernous-carotid (C-C) fistulas occur as a consequence of an abnormal communication between the carotid artery and the cavernous sinus. The high-pressure venous state caused by transfer of arterial pressure results in a backup of flow, with resultant examination findings of proptosis, chemosis, conjunctival injection, tortuous ‘corkscrew’ vessels and possibly ophthalmoparesis (figure 9).56 C-C fistulas are best diagnosed with a conventional angiogram, but an enlarged ophthalmic vein can be seen with standard radiographic imaging in some cases.
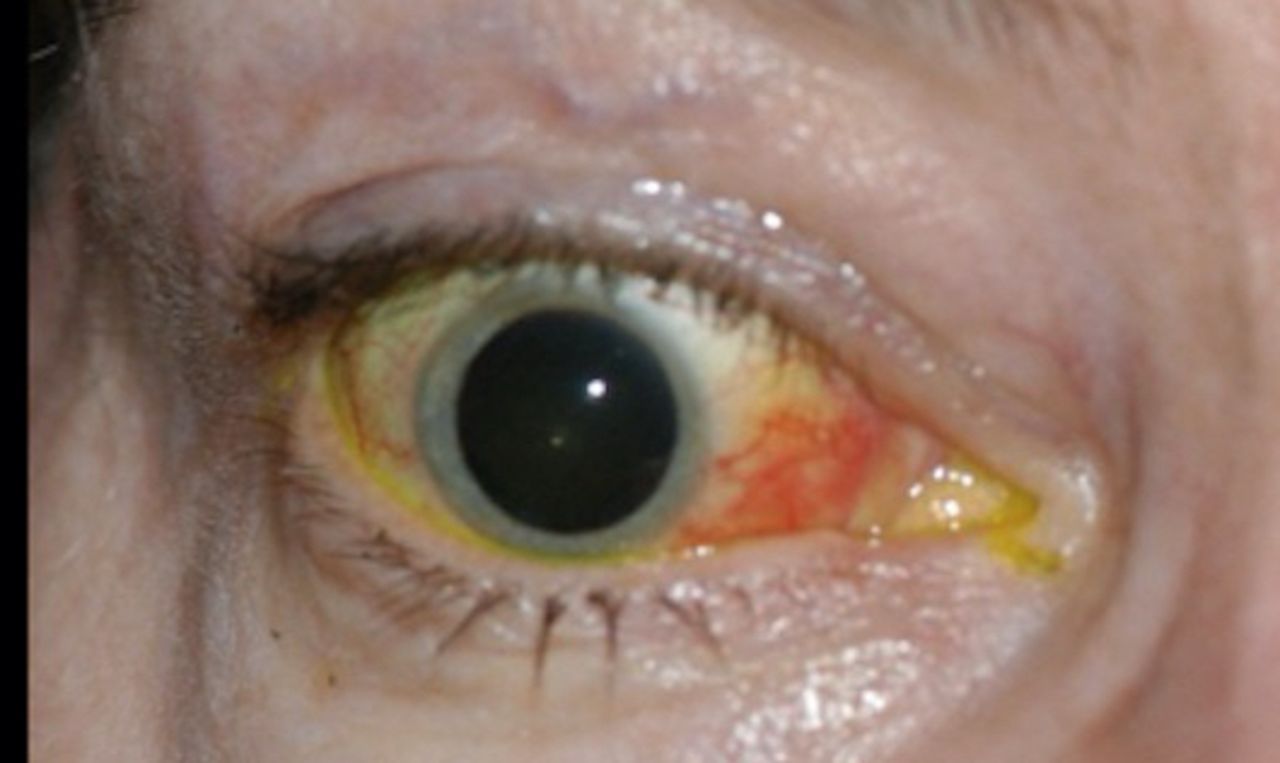
Ocular sequela of cavernous-carotid fistula. Right eye of a patient with a cavernous-carotid fistula showing proptosis, chemosis and conjunctival injection.
In general, the anatomy of many of the structures described above can be reviewed radiographically (figure 10). Besides those vascular causes of vision disturbances reviewed above, the physician should recognise that stroke mimics can also involve the visual system. For example, migraine can cause transient vision disturbance similar to AF. Migraine vision disturbance often lasts longer, and the visual symptoms are positive (flashing lights, scintillations). Demyelinating disorders such as multiple sclerosis can involve vision, such as with optic neuritis. Unlike ischaemic optic neuropathy, the vision loss in optic neuritis almost always resolves over time, and pain on eye movements is a more prominent symptom. Finally, temporal arteritis can cause vision loss due to inflammatory occlusion of the disc head blood supply. Patients with giant cell arteritis are older, have headaches and jaw claudication and present with elevated sedimentation rates.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
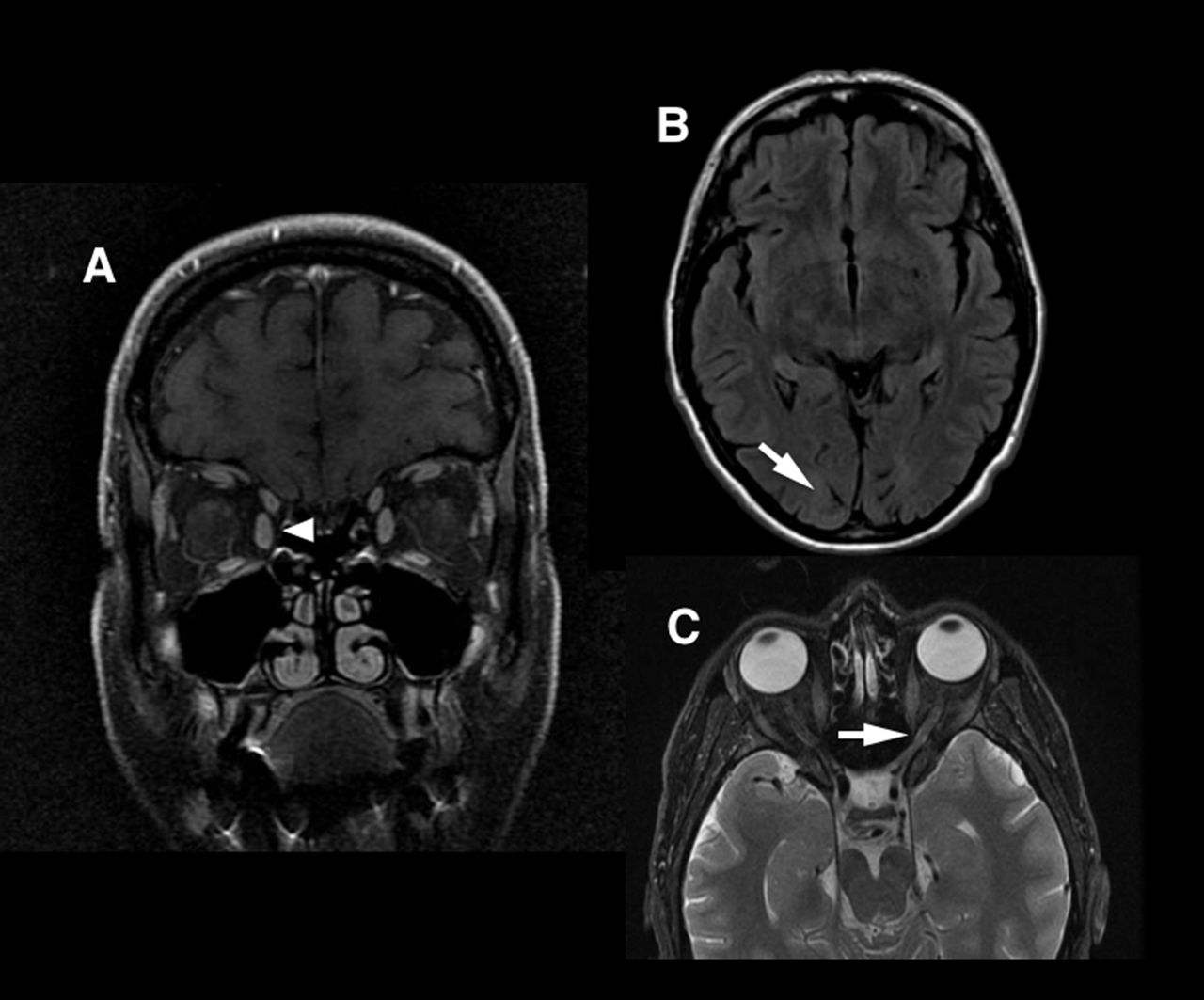{kind=link}
Anatomy of visual structures on MRI. (A) Coronal orbital view (arrowhead at medial rectus muscle). (B) Axial FLAIR MRI (arrow at occipital pole). (C) Axial fat-saturated orbital sequence (arrow at optic nerve).
Conclusion
There is no singular presentation of visual dysfunction in stroke. A small occipital pole embolus can cause a complete hemianopia and cut-off a person from his independence with loss of driving privileges. Bilateral posterior infarctions resulting in Anton’s Syndrome result in total blindness, yet the patient will not recognise the vision loss. Vision problems are not as widely recognised as motor or speech defects as a sign of stroke, and treatments remain limited both for acute and chronic therapy after stroke-related vision loss. Hence, continued education, study and investigation of the visual aspects of strokes in paramount for the present and future health of these patients.
Footnotes
Contributors JHP has written half of the manuscript, performed the proofing, researched the topics and approved the final manuscript. CAY has also written half of the manuscript as well as researching the topics and providing a literature review and approved the final manuscript.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Commissioned; externally peer reviewed.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
References
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.
- 40.
- 41.
- 42.
- 43.
- 44.
- 45.
- 46.
- 47.
- 48.
- 49.
- 50.
- 51.
- 52.
- 53.
- 54.
- 55.
- 56.